1. Introduction
Crigler-Najjar syndrome type I (CN-I; MIM #218800) is the most severe type of hereditary unconjugated hyperbilirubinemia, a disease first recognized by Crigler and Najjar (
1952). CN-I is inherited as an autosomal recessive condition with an estimated frequency of less than one per million live births (Petit et al.,
2008). The clinical manifestation of CN-I is severe icterus. The serum bilirubin concentration associated with CN-I is greater than 20 mg/dl (343 mmol/L), even 30–50 mg/dl (513–855 mmol/L), and the bilirubin is entirely unconjugated (Maruo et al.,
2011). Phenobarbital treatment is not effective for the treatment of symptoms. Patients with CN-I are in danger of kernicterus and must receive daily phototherapy immediately in the neonatal period. Liver transplantation is eventually required.
CN-I is caused by homozygous or compound heterozygous mutations of the UDP-glycuronosyl-transferase gene (
UGT1A1; MIM #191740) on chromosome 2q37 (Costa et al.,
2009), leading to a complete and non-inductile deficiency of bilirubin UDP-glucuronosyltransferase 1-1 (UGT1A1) activity. UGT1A1 (ENSP00000304845) is a membrane enzyme of the endoplasmic reticulum comprising 533 amino acids, and is expressed only in the liver, esophagus, stomach, and intestine (Tukey and Strassburg,
2001; Basu et al.,
2004). The signal peptide of UGT1A1 is composed of amino acids 1–27; amino acids 29–468 constitute the superfamily domains; and, the transmembrane region is composed of amino acids 13–35 and 491–513 (Wilke et al.,
2000). UGT1A1 plays a critical role in the bilirubin-conjugation process (Tukey and Strassburg,
2001).
The first mutation described in
UGT1A1 in 1992 was a nonsense mutation found in a homozygous state in a patient with CN-I (Bosma et al.,
1995). To date, more than 130 mutations have been identified, most of which are missense or nonsense mutations (Canu et al.,
2013).
In this paper, we studied two CN-I patients from two unrelated families. Three mutations were identified: c.239_245delCTGTGCC (p.Pro80HisfsX6), c.1253delT (p.Met418ArgfsX5), and c.1156G>T (p.Val386Phe) of
UGT1A1. Moreove, we confirmed that one patient had an entire deletion on one allele of the
UGT1A1 gene.
2. Materials and methods
2.1. Patient A
Patient A was 3.5 months old, a Chinese female of nonconsanguineous parents. She was the fifth fetus and the second newborn baby of her mother; the first three fetuses were voluntarily aborted, and the fourth fetus was her older deceased sister. Patient A was born at full term (weight 3600 g) by normal vaginal delivery following a normal pregnancy. At 3 d after birth, she began to demonstrate icterus; at 7 d, her serum total bilirubin level was 570.5 μmol/L and unconjugated bilirubin level was 549.9 μmol/L, and she was admitted to the local hospital. She was treated with daily phototherapy, albumin infusion, and exchange blood transfusion. Upon treatment, her serum total bilirubin decreased to 169.2 μmol/L. Soon after the treatment, the bilirubin level increased. Yinzhihuang oral liquid was administered to decrease her bilirubin, but no effect was observed. She was subsequently admitted to the Affiliated Children’s Hospital of Capital Institute of Pediatrics (Beijing, China). Upon admission, the physical examination revealed severe icterus, a body weight of 9200 g, a head circumference of 42.5 cm, normal neuromotor development, and no signs of kernicterus. Laboratory findings were as follows: serum total bilirubin, 589.5 μmol/L (3.4–20.0 μmol/L) (Olympus AU640, Japan); serum unconjugated bilirubin, 571.8 μmol/L; alanine aminotransferase, 41.2 U/L (0–40 U/L); aspartate aminotransferase, 34.8 U/L (0–40 U/L); hemoglobin, 114 g/L (110–190 g/L); and, reticulocyte count, 1.1% (0.5%–1.5%). Treatment with phenobarbital intramuscular injection for two days (4 mg/kg per day) did not reduce her serum bilirubin levels; therefore, the patient was discharged. The patient’s outcome was unknown because of loss to follow-up. The serum bilirubin concentrations of the patient’s parents were not measured, and they did not demonstrate jaundice; however, the patient’s sister had severe hyperbilirubinemia after birth, experienced convulsions at 7 months, and died of kernicterus at 22 months.
2.2. Patient B
Patient B was 4.5 months old, a Chinese male of nonconsanguineous parents. He was the first child of the family. He was born at full term (weight 3200 g) by normal vaginal delivery, but his mother had been considering abortion during the pregnancy. Although his parents displayed no apparent jaundice, patient B appeared to be jaundiced at 6 d after birth and then developed severe icterus. At one month, his serum total bilirubin was 411.6 μmol/L and unconjugated bilirubin was 403.1 μmol/L; he was admitted to the local hospital and treated daily with phototherapy. Upon treatment, the transcutaneous bilirubin decreased slightly (to 16.5 mg/dl). Bilirubin levels increased immediately when phototherapy stopped, and Yinzhihuang oral liquid was administered, but no significant reduction in bilirubin was observed. At four months of age, his serum total bilirubin was 520.6 μmol/L and unconjugated bilirubin was 499.6 μmol/L, and he was transferred to the Affiliated Children’s Hospital of Capital Institute of Pediatrics (Beijing, China) for further treatment. Upon admission, the physical examination revealed severe icterus, a body weight of 7000 g, a height of 66 cm, a head circumference of 41 cm, a slight retardation in mental and motor development (he could not raise his head), and no history of somnolence, vomit, convulsions, or opisthotonos. Laboratory findings were as follows: serum total bilirubin, 496.5 μmol/L (Olympus AU640, Japan); serum unconjugated bilirubin, 490.7 μmol/L; alanine aminotransferase, 34.1 U/L; aspartate aminotransferase, 21.8 U/L; and, hemoglobin, 111 g/L. Treatment with oral-phenobarbital for 8 d (10 mg/kg per day) did not reduce his serum bilirubin levels. At 13 months of age, patient B died of kernicterus.
2.3. DNA sequencing
Genomic DNA was isolated from the peripheral blood leukocytes of the patients and their parents, using the phenol-chloroform method (PC) (Sambrook et al.,
1989), with their informed consent. Five exons and their flanking sequences, specifically the promoter region of
UGT1A1, were amplified with primers as described by Maruo et al. (
1999). Then, direct sequencing of purified polymerase chain reaction (PCR) products was performed using an ABI 3700 automated sequencer (Applied Biosystems, USA).
2.4. Determination of UGT1A1 gene copy number by quantitative real-time PCR (qRT-PCR)
Primers (Table
1) were designed using Primer Express 3.0 software (Applied Biosysems) for qRT-PCR. P3–P8 were in
UGT1A1. Primers of P1, P2, and P9 were designed to determine the deletion range of patient B. qRT-PCR was carried out using the ABI Prism™ 7500 sequence detection system and UltraSYBR Mixture (with ROX) (CW0956A, CWbio Co., Ltd., China) in reaction volumes of 25 μl. All reactions of the same run were prepared from the same master mix (2× UltraSYBR Mixture, 12.5 μl; forward primer, 0.5 μl; reverse primer, 0.5 μl; DNA, 50 ng; add high performance liquid chromatography (HPLC)-pure water to 25 μl). Each pair of test and control samples was amplified in three different wells. Because there was no positive control, we conducted only a normal control. The PCR conditions were as follows: 10 min, 95 °C; 40 cycles of 15 s at 95 °C and 1 min at 62 °C; and, run for about 2 h (Wilke et al.,
2000).
Table 1
Primers for qRT-PCR
| Gene |
Primer |
Primer sequence (5′–3′) |
Product size (bp) |
|
USP40
|
P1 |
F: GCCTTTGCCTTCTTCCCTTT |
140 |
| R: GAAGTAGCCGGCAACCAAAA |
| Upstream of UGT1A8
|
P2 |
F: AAGGCCTCTCAGGGTTTGGA |
102 |
| R: GCCCTCAGCACTGACTTCCT |
|
UGT1A1
|
P3 (PBREM) |
F: CCGGACTTGGCACTTGGTAA |
130 |
| R: GCCATTCTGGATCCCTTGCT |
| P4 (Exon 1) |
F: ACGTACCCTGTGCCATTCCA |
100 |
| R: TGATCACACGCTGCAGGAAA |
| P5 (Exon 2) |
F: GCTGATGCTTTGGGCAAAA |
85 |
| R: GGGATTAGCGCTCCTGTGAA |
| P6 (Exon 3) |
F: AGGTCCTGTGGCGGTACACT |
110 |
| R: ATCCAATCCGCCCAACATAC |
| P7 (Exon 4) |
F: CGATGACCCGTGCCTTTATC |
118 |
| R: CGCTTTGCATTGTCCATCTG |
| P8 (Exon 5) |
F: GCTATGAAGTGCTGGGCAAGT |
110 |
| R: AAATGCGACACCAGTGAAAACA |
|
TRPM8
|
P9 |
F: CCAGGCTCAGCATGAGGAA |
115 |
| R: TGCGCATGACTTACGCTTTC |
|
GAPDH
|
Exon 4 |
F: AAGGCTGAGAACGGGAAGCT |
105 |
| R: CCTAGTTGCCTCCCCAAAGC |
Data evaluation was carried out using the ABI Prism™ 7500 sequence detection software. We used the ΔΔ
C
T method to analyze the gene copy number compared with that of healthy samples. The 2
−ΔΔC
T
was the ratio of inspected samples to controlled samples, specifically the ratio of target gene copy number to
GAPDH copy number.
3. Results
3.1. Gene mutations in patient A and her parents
Mutations of the
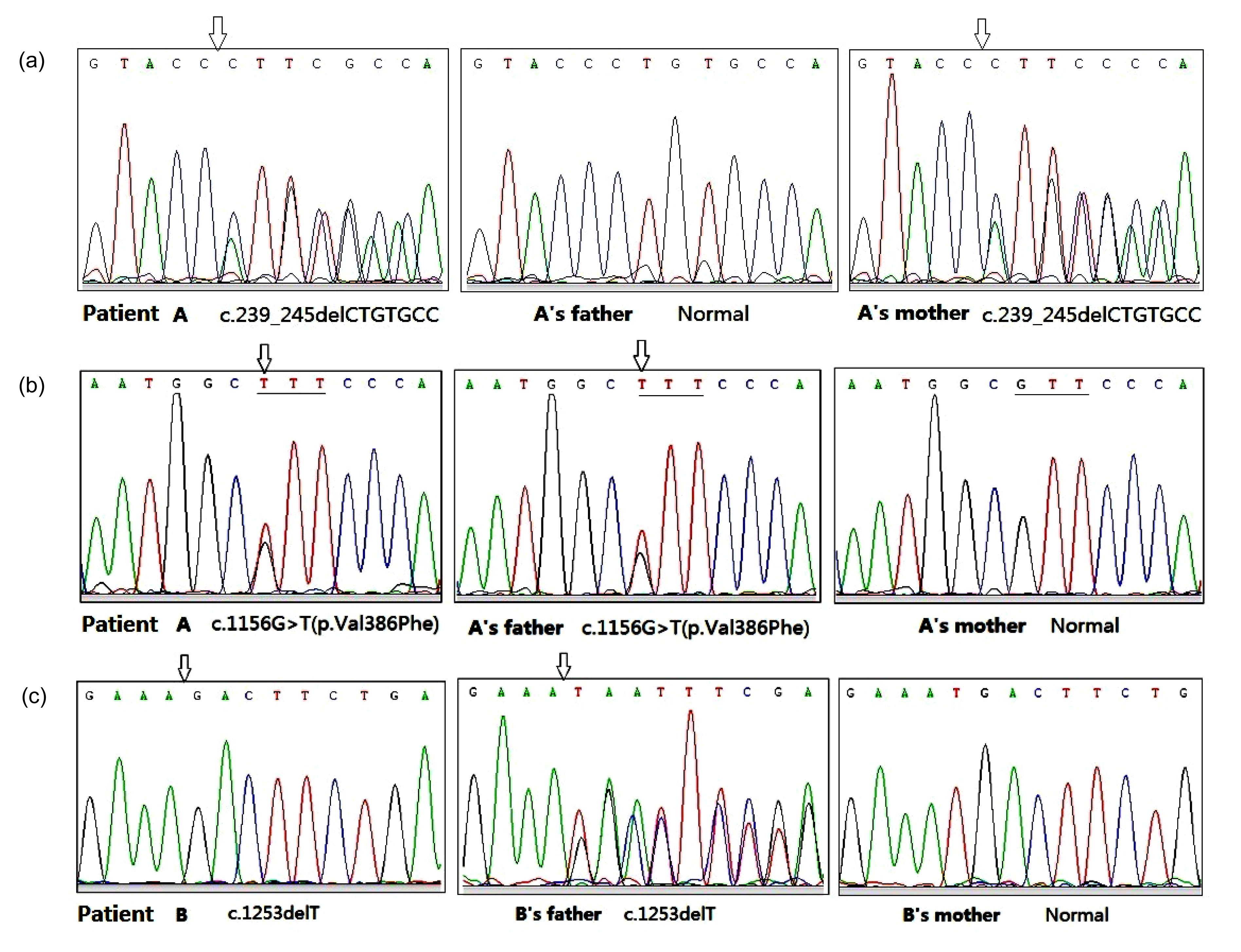
UGT1A1 gene in patient A were c.239_245delCTGTGCC and c.1156G>T (Figs.
1a and 1b). The two mutations were observed in her parents, respectively, and therefore, were not spontaneous. Mutation c.239_245delCTGTGCC was predicted to cause an 85-amino acid truncated protein. To analyze the second mutation, c.1156G>T (p.Val386Phe), we first predicted this substitution base in ESEfinder Release 2.0 (
http://rulai.cshl.edu/tools/ESE2/). We found that one default threshold value of wild sequence (CGTTCCCA) bound with SC35 protein was 2.529. However, this mutant sequence (1156G>T) (CTTTCCCA) was defective. When the default threshold of SC35 protein is above 2.383, the 8-base sequence can bind with SC35 protein. This indicated that this missense mutation was meaningful and may affect RNA splicing by affecting binding with SC35. Then we compared the 30-amino acid residues 371–400 of UGT1A1 from ten species, and found that Val-386 of the human UGT1A1 was highly conserved and, therefore, likely to be an important amino acid. We also predicted mutation p.Val386Phe in SIFT (sorting intolerant from tolerant) and Polyphen software online, and the resulting scores were 0 (damaging) and 0.995 (deleterious). In addition, we searched c.1156G>T in the 1000 Genomes Project (
http://www.1000genomes.org/), and found that this mutation was not a known polymorphic site.
Fig.1
Mutations of UGT1A1 in the two patients
(a) In patient A: the deletion mutation c.239_245delCTGTGCC was found in patient A and her mother, and both were heterozygous; her father was normal at c.239_245. (b) In patient A: the missense mutation c.1156G>T (p.Val386Phe) was found in patient A and her father; her mother was normal at the same site. (c) In patient B: mutation c.1253delT in patient B was a homozygous deletion; his father had a heterozygous deletion; his mother was normal and had no base variation at the same site
3.2. Gene mutations in patient B and his parents
The mutation of the
UGT1A1 gene in patient B was c.1253delT, which looked from sequencing like a homozygous deletion. His father had a heterozygous deletion, but no base change was found at the same cDNA site in his mother (Fig.
1c). To explain the sequencing results, we postulated that there was a
UGT1A1 gene deletion in patient B’s mother, and that this gene deletion was inherited by patient B. qRT-PCR was performed to verify our postulate (Fig.
2).
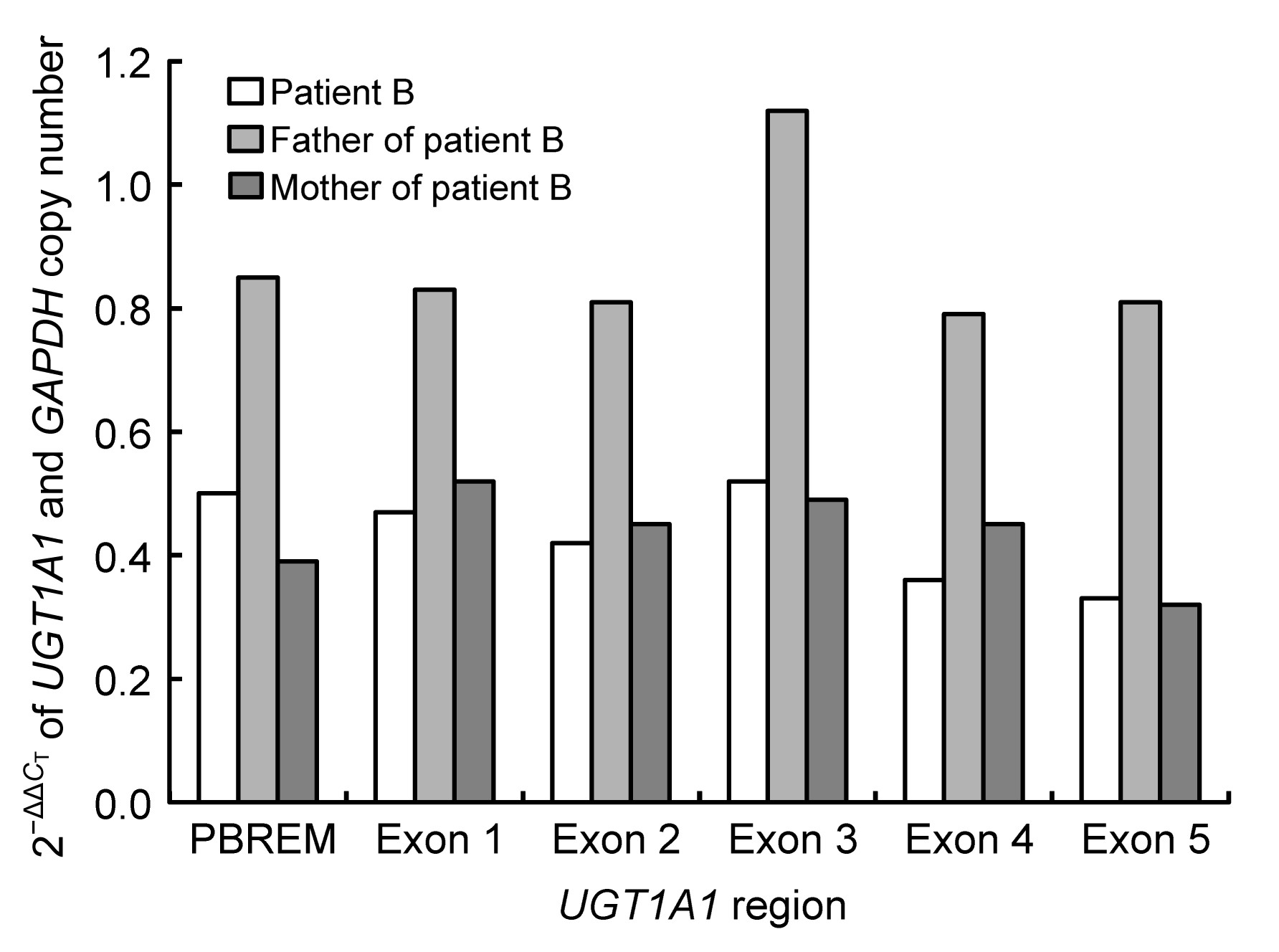
Fig.2
qRT-PCR results for UGT1A1 2−ΔΔC
T
in family B
Patient B: 0.32–0.52; his mother: 0.32–0.52; his father: 0.79–1.12
The 2
−ΔΔC
T
value of the normal control was considered to be 1. The father’s values were between 0.79 and 1.12, similar to that of the control. The 2
−ΔΔC
T
values of patient B and his mother were similar; their values were both between 0.32 and 0.52 from the upstream phenobarbital-responsive enhancer module (PBREM) region to exon 5, and were about half of the father’s value. These data indicate that patient B and his mother had only one copy of the
UGT1A1 gene, i.e., they were hemizygous. Therefore, in patient B, one allele with an entire
UGT1A1 deletion was maternally inherited, and the other with mutation 1253delT was paternally inherited.
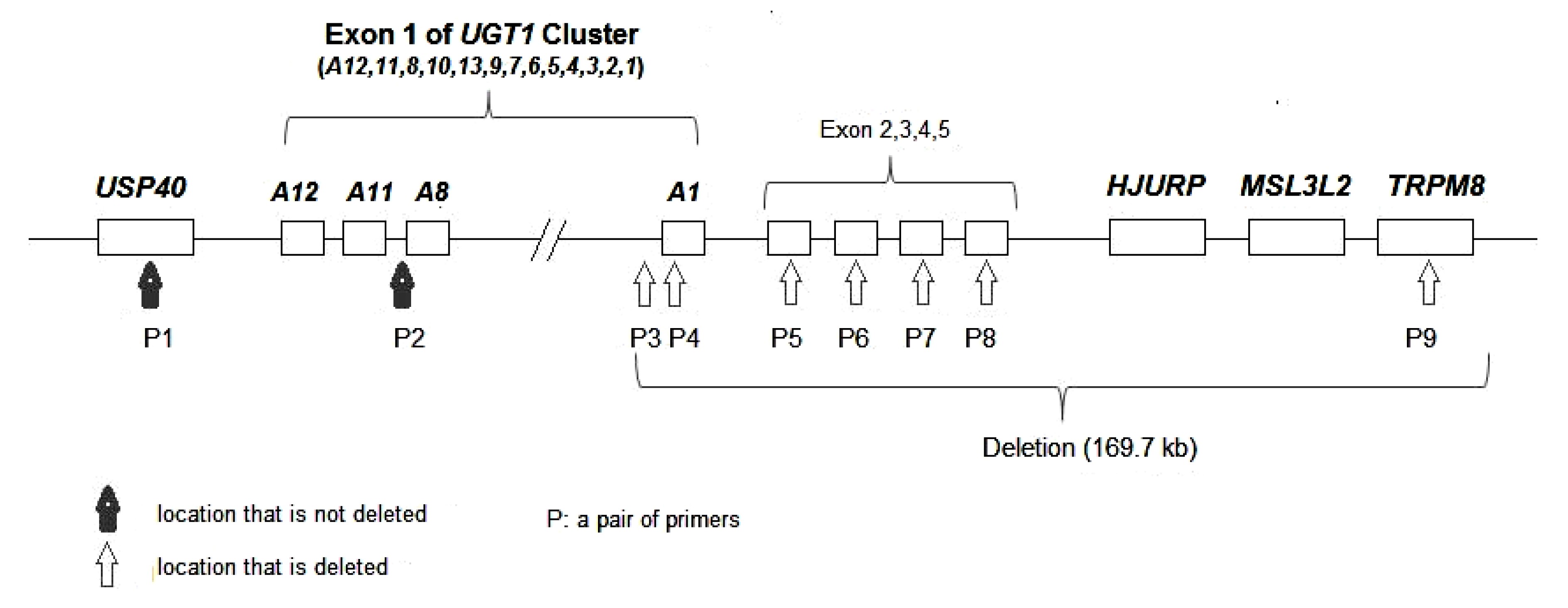
qRT-PCR results of three other regions (
USP40 and upstream of
UGT1A8 and
TRPM8) showed that the
USP40 and upstream region of
UGT1A8 had two copies (1.00 and 1.01) and
TRPM8 had only one copy (0.47). Thus, we could speculate that the deletion extended at least 169.7 kb from the PBREM region of
UGT1A1 to
TRPM8 (Fig.
3).
Fig.3
Locations of primers and a draft genomic deletion map encompassing the UGT1A1 gene and flanking regions in patient B
The maternal chromosome 2 from P3 to P9 is deleted; P1 and P2 are not deleted. The length of the deletion from the PBREM region of UGT1A1 to TRPM8 is 169.7 kb
Searches of the databases of National Center for Biotechnology Information (NCBI), Ensembl, and Human Gene Mutation Database (HGMD) up to October 2013 showed no reports of mutations c.239_245delCTGTGCC and c.1253delT. The main clinical manifestations and molecular analyses of the two CN-I patients are summarized in Tables
2 and
3, respectively.
Table 2
Main clinical manifestations of the two CN-I patients
| Patient |
Gender |
Age (month) |
Kernicterus |
Serum bilirubin (μmol/L)
|
Phenobarbital treatment |
Outcome |
| Total |
Unconjugated |
| A |
Female |
3.5 |
No |
589.5 |
571.8 |
Not effective |
Unknown |
| B |
Male |
4.5 |
Uncertain |
496.5 |
490.7 |
Not effective |
Died of kernicterus at 13 months |
Table 3
Molecular analyses of the two CN-I patients
| Patient |
UGT1A1 gene |
cDNA change |
Amino acid change |
Location |
Mutation type |
Source |
(TA) polymorphism |
| A |
Allele 1 |
c.1156G>T |
p.Val386Phe |
Exon 4 |
Missense |
Paternal |
(TA)6
|
| Allele 2 |
c.237_243del CTGTGCC |
p.Pro80HisfsX6*
|
Exon 1 |
Frame shift |
Maternal |
(TA)7
|
| B |
Allele 1 |
c.1253delT*
|
p.Met418ArgfsX5*
|
Exon 4 |
Frame shift |
Paternal |
(TA)6
|
| Allele 2 |
Deletion of whole gene |
Maternal |
*Novel mutation of UGT1A1
4. Discussion
For patient A, RNA from mutation c.239_245delCTGTGCC was probably degraded by an RNA decay phenomenon, and a functional protein was also probably deficient.
On the other hand, a truncated protein of 85 amino acids (p.Pro80HisfsX6) was translated by RNA from this mutation, but lost most of the functional domain (p.86–p.533) of the UGT1A1 enzyme. Clearly, this allele of
UGT1A1 with mutation c.239_245delCTGTGCC had lost gene function. The other allelic mutation, Phe-386 (c.1156G>T), substitutes the highly conserved Val-386 of UGT1A1. To predict its pathogenic mechanism, we conducted predictions of RNA splicing and protein function. First, we found that this missense mutation might affect RNA splicing by affecting binding with SC35 protein. However, it has been reported that
UGT1A1 is expressed only in the liver, esophagus, stomach, and intestine (Tukey and Strassburg,
2001; Basu et al.,
2004). So we could not verify this effect by trial. Predictions at the protein level indicated that most amino acids from 370 to 400 were highly conserved. Previous studies indicated that p.Ser376Phe (c.1124C>T), p.Ser381Arg (c.1143C>G), and p.Pro387Ala or p.Pro387lLeu (c.1159C>G, c.1160C>T) mutations resulted in an inactive UGT1A1 enzyme (Kadakol et al.,
2000; Sneitz et al.,
2010). This showed that the region from 370 to 400 amino acids is important.
On the other hand, the SIFT (0) and Polyphen (0.995) scores showed that the p.Val386Phe missense mutation was deleterious. However, a known missense mutation at this site, p.Val386Ile, found in Gilbert syndrome (Canu et al.,
2013), with a SIFT score of 0.210 and a Polyphen score of 0.665, does not lead to deleterious symptoms. According to clinical results and software prediction data, mutation p.Val386Phe is more deleterious than p.Val386Ile. Valine and isoleucine are aliphatic, neutral, hydrophobic amino acids, while phenylalanine is an aromatic, neutral, hydrophobic amino acid. While p.Val386Ile likely affects the enzyme activity, perhaps it can be tolerated, because isoleucine and valine have similar structures. Although the properties of valine and phenylalanine are similar, steric hindrance from the large phenyl side chain of Phe, may affect the secondary structures of the protein. Petit et al. (
2004) identified a CN-I patient with homozygous p.Val386Phe, and also indicated that p.Val386Phe was damaging. In conclusion, we believe that the missense mutation p.Val386Phe is likely to result in an inactive enzyme on account of the manifestation of severe icterus, non-effective phenobarbital treatment, prediction results, and a truncated inactive enzyme formed by the other allele. However, the pathogenic mechanism of the p.Val386Phe mutation remains to be determined.
For patient B, the maternal
UGT1A1 allele was deleted entirely. A 422-amino acid truncated protein (p.Met418ArgfsX5) was predicted as a result of the c.1253delT mutation of the paternal allele, lacking 111 amino acids of the UGT1A1 C-terminus, and predicted to lack the transmembrane domain. Also, abnormal RNA produced by mutation c.1253delT was probably degraded by an RNA decay pathway. Because patient B had severe icterus, phenobarbital treatment was not effective, and he died of kernicterus at 13 months, we concluded that the paternal allele with the c.1253delT mutation had lost its function. Because sequencing results did not completely explain the base variation at c.1253delT found in family B, we explored and confirmed the possibility of an allelic deletion of the
UGT1A1 gene. The length of the deletion of patient B’s maternal chromosome 2 was speculated to be at least 169.7 kb. To our knowledge, entire allelic deletions identified in CN-I patients have rarely been reported.
The two CN-I patients in this study were treated with daily phototherapy, albumin infusion, and exchange transfusion. These therapies temporarily decreased bilirubin levels. As patients were hospitalized for only several days, some more effective therapies, such as liver transplantation and hepatocyte transplantation (Lucey et al.,
2000; Lysy et al.,
2008), were not applied. In the near future, gene therapy may be a prospective therapeutic method (Bortolussi et al.,
2012).
In summary, we identified three mutations, c.239_245delCTGTGCC (p.Pro80HisfsX6), c.1253delT (p.Met418ArgfsX5), and c.1156G>T (p.Val386Phe), and an entire
UGT1A1 gene deletion, in two CN-I patients. The former two mutations were novel and were considered as pathogenic mutations. While software predictions showed that the third mutation, c.1156G>T, affects RNA splicing and protein function, its pathogenic mechanism remains unknown. We also confirmed one patient had lost heterozygosity of the
UGT1A1 gene by inheriting a deletion of one entire allele.
Acknowledgements
We would like to thank the two patients and their family members for their cooperation in this study.
Compliance with ethics guidelines Yan LI, Yu-jin QU, Xue-mei ZHONG, Yan-yan CAO, Li-min JIN, Jin-li BAI, Xin MA, Yu-wei JIN, Hong WANG, Yan-ling ZHANG, and Fang SONG declare that they have no conflict of interest.References
[1] Basu, N.K., Ciotti, M., Hwang, M.S., 2004. Differential and special properties of the major human UGT1-encoded gastrointestinal UDP-glucuronosyltransferases enhance potential to control chemical uptake.
J Biol Chem, 279(2):1429-1441.


[2] Bortolussi, G., Zentilin, L., Baj, G., 2012. Rescue of bilirubin-induced neonatal lethality in a mouse model of Crigler-Najjar syndrome type I by AAV9-mediated gene transfer.
FASEB J, 26(3):1052-1063.
[3] Bosma, P.J., Chowdhury, J.R., Bakker, C., 1995. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome.
N Engl J Med, 333(18):1171-1175.
[4] Canu, G., Minucci, A., Zuppi, C., 2013. Gilbert and Crigler-Najjar syndromes: an update of the UDP-glucuronosyltransferase 1A1 (
UGT1A1) gene mutation database.
Blood Cells Mol Dis, 50(4):273-280.
[5] Costa, E.V., Vieira, E., Lopes, A.I., 2009. Identification of a novel deletion in UDP-glucuronosyltransferase gene in a patient with Crigler-Najjar syndrome type I.
Blood Cells Mol Dis, 42(3):265-266.
[6] Crigler, J.F., Najjar, V.A., 1952. Congenital familial nonhemolytic jaundice with kernicterus.
Pediatrics, 10(2):169-180.
[7] Kadakol, A., Ghosh, S.S., Sappal, B.S., 2000. Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase (
UGT1A1) causing Crigler-Najjar and gilbert syndromes: correlation of genotype to phenotype.
Hum Mutat, 16(4):297-306.
[8] Lucey, J.F., Suresh, G.K., Kappas, A., 2000. Crigler-Najjar syndrome, 1952–2000: learning from parents and patients about a very rare disease and using the Internet to recruit patients for studies.
Pediatrics, 105(5):1152-1153.
[9] Lysy, P.A., Najimi, M., Stephenne, X., 2008. Liver cell transplantation for Crigler-Najjar syndrome type I: update and perspectives.
World J Gstroenterol, 14(22):3464-3470.
[10] Maruo, Y., Nishizawa, K., Sato, H., 1999. Association of neonatal hyperbilirubinemia with bilirubin UDP-glucuronosyltransferase polymorphism.
Pediatrics, 103(6):1224-1227.
[11] Maruo, Y., Ozgenc, F., Mimura, Y., 2011. Compound heterozygote of a novel missense mutation (p.K402T) and a double missense mutation (p.[G71R;Y486D]) in type II Crigler-Najjar syndrome.
J Pediatr Gastroenterol Nutr, 52(3):362-365.
[12] Petit, F.M., Gajdos, V., Francoual, J., 2004. Allelic heterogeneity of Crigler-Najjar type I syndrome: a study of 24 cases.
Clin Genet, 66(6):571-572.
[13] Petit, F.M., Bzieau, S., Gajdos, V., 2008. The Tunisian population history through the Crigler-Najjar type I syndrome.
Eur J Hum Genet, 16(7):848-853.
[14] Sambrook, J., Fitsch, E.F., Maniatis, T., 1989. Molecular Cloning: A Laboratory Manual+Cold Spring Harbor. Cold Spring Harbor Laboratory Press,NY, USA :
[15] Sneitz, N., Bakker, C.T., de Knegt, R.J., 2010. Crigler-Najjar syndrome in the Netherlands: identification of four novel
UGT1A1 alleles, genotype-phenotype correlation, and functional analysis of 10 missense mutants.
Hum Mutat, 31(1):52-59.
[16] Tukey, R.H., Strassburg, C.P., 2001. Genetic multiplicity of the human UDP-glucuronosyltransferases and regulation in the gastrointestinal tract.
Mol Pharmacol, 59(3):405-414.
[17] Wilke, K., Duman, B., Horst, J., 2000. Diagnosis of haploidy and triploidy based on measurement of gene copy number by real-time PCR.
Hum Mutat, 16(5):431-436.
Open peer comments: Debate/Discuss/Question/Opinion
<1>