1. Introduction
Nance-Horan syndrome (NHS) (OMIM: 302350) is a rare X-linked syndrome, first reported in detail by Horan and Billson (
1974) and Nance et al. (
1974). Affected male patients often manifest bilateral congenital dense nuclear cataracts, characteristic dental anomalies, craniofacial abnormalities, and, in some cases, mental retardation (MR) (Toutain et al.,
1997; Ding et al.,
2009). Congenital dense nuclear cataracts in affected males usually lead to profound vision loss; thus, cataract surgery should be performed at an early age (Burdon et al.,
2003). Other ophthalmological features include microcornea, microphthalmia, nystagmus, and strabismus. Dental abnormalities characteristic of NHS consist of screwdriver-shaped incisors, supernumerary maxillary incisors (mesiodens), and diastema (Walpole et al.,
1990). Heterozygous females display similar, but milder clinical manifestations than affected males (Ding et al.,
2009).
NHS is caused by mutations in the
NHS gene, which comprises 10 coding exons and encodes at least 5 isoforms as a result of alternative splicing (Brooks et al.,
2010). NHS-A and NHS-1A are two major isoforms of the NHS protein. The two isoforms are transcribed from exon 1 of the
NHS gene, encoding a 1630-amino-acid protein and a 1651-amino-acid protein, respectively (Brooks et al.,
2010). The
NHS gene is expressed in the midbrain, lens, tooth, retina, and dental primordia, in a spatially and temporally regulated pattern, and is highly conserved across vertebrate species, such as mouse, rat, and zebrafish (Huang et al.,
2007; Sharma et al.,
2008).
To date, approximately 27 mutations in the
NHS gene have been identified (Burdon et al.,
2003; Brooks et al.,
2004; Ramprasad et al.,
2005; Florijn et al.,
2006; Huang et al.,
2007; Sharma et al.,
2008; Coccia et al.,
2009; Chograni et al.,
2011; Khan et al.,
2012; Tug et al.,
2013), including nonsense mutations, frameshift mutations, genomic rearrangements, and one missense mutation. In this study, we identified a novel nonsense mutation c.322G>T (E108X) in the
NHS gene’s exon 1 in a family with NHS by a combination of exome sequencing and Sanger sequencing. The clinical features in all affected males and female carriers are described in detail.
2. Materials and methods
2.1. Subjects and ethics approval
This study followed the tenets of the Declaration of Helsinki and was approved by the Ethics Committee of the First Affiliated Hospital, School of Medicine, Zhejiang University, China. A three-generation family with congenital cataracts, microcornea, strabismus, and nystagmus was recruited. Seven members of the family participated in the study, three affected (II:1, II:2, and III:1) and four unaffected (I:1, I:2, I:3, and II:3) members. Informed consent and blood samples were obtained from all the participants in the family. All participants underwent comprehensive ophthalmological examinations including visual acuity, slit-lamp biomicroscopy, fundus examination, intraocular pressure (IOP) measurement, and IOLMaster testing. Nonocular features related to NHS were also noted. A total of 50 healthy individuals without congenital ocular diseases were selected serving as an ethnically-matched control. Genomic DNA was extracted using the QIAamp DNA Blood Midi kit (Qiagen, Hilden, Germany), according to the manufacturer’s protocol.
2.2. Exome sequencing
One affected male (III:1) in the family was selected for exome sequencing. Exome sequencing was carried out using the Agilent SureSelect Human All Exon v4.0 (51M) kit, according to the manufacturer’s protocol. The 3 μg of genomic deoxyribonucleic acid (DNA) was randomly fragmented into pieces 200–300 bp in size using a Covaris acoustic system, followed by end-repair, A-tailing, and Covaris acoustic measurements. The adapter-ligated templates were purified using Agencourt AMPure SPRI beads and amplified by four-cycle ligation-mediated polymerase chain reaction (LM-PCR), under the following PCR condition: 2 min at 94 °C, four cycles of 10 s at 94 °C, 30 s at 62 °C, and 30 s at 72 °C, then 300 s at 72 °C. The LM-PCR products were hybridized to the Agilent Oligo pool for enrichment for 24 h at 65 °C. Hybridized fragments were bound to streptavidin beads and non-hybridized fragments were washed out. Captured LM-PCR products were amplified by PCR (2 min at 98 °C; 10–12 cycles for 10 s at 98 °C, 30 s at 60 °C, 30 s at 72 °C, followed by 300 s at 72 °C). The magnitude of enrichment of the samples was estimated with an Agilent 2100 bioanalyzer and quantitative PCR. The captured library was sequenced on Illumina HiSeq2000 analyzers for 90 cycles per read, to generate paired-end reads and 8 bp of the index tag (following the manufacturer’s standard sequencing protocols).
2.3. Mapping, single-nucleotide polymorphism (SNP), and insertion and deletion (InDel) calling
Image analysis and base calling were performed according to the Illumina pipeline. Indexed primers were used for data fidelity surveillance. Short Oligonucleotide Analysis Package (SOAP) aligner (Ver. 2.21) was used to map the clean reads to the human reference genome (hg19) with a maximum fault-tolerance of 3-bp mismatches. Based on these results, the SOAPsnp software (Ver. 1.05) was used to assemble the consensus sequence and call genotypes in target regions. Candidate single-nucleotide SNPs were selected using the following criteria: SNP quality ≥20, sequencing depth ≥4-fold, estimated copy number ≤2, and distance between two SNPs >5 bp. For small InDels <20 bp in exome regions, we mapped the reads to the reference genome with the Burrows Wheeler Aligner (BWA; Ver. 0.6.2), and passed the result to the Genome Analysis Tool kit (GATK) to identify breakpoints.
2.4. PCR and Sanger sequencing
Following the results of exome sequencing, Sanger sequencing was performed to verify the genetic defects. Primers flanking the mutation area of
NHS (NM_198270) were designed, based on the reference genomic sequences of the human genome from GenBank at the National Center for Biological Information (NCBI), and synthesized by Invitrogen, Shanghai, China. The forward primer was 5'-TTCGCCAAGCGGATCGTGGA-3', and the reverse primer was 5'-TTAGGGTCAAGCGTGCTGAGGA-3'. PCR amplification was carried out using an Applied Biosystems Inc. (ABI) 9700 Thermal Cycler. PCR products were directly sequenced on an ABI PRISM 3730 automated sequencer (Applied Biosystems). Sequence data comparisons and analyses were performed using DNASTAR SeqMan. Co-segregation analysis was subsequently performed on the DNA samples from available family members.
2.5. Bioinformatics analysis
Multiple sequence alignment of NHS protein sequences from
Homo sapiens (GenPept accession NP_938011.1),
Mus musculus (GenPept accession NP_001074521.1),
Bos taurus (GenPept accession NP_001192657.1),
Macaca mulatta (GenPept accession XP_001104229.2),
Canis lupus familiaris (GenPept accession XP_548877.2),
Sus scrofa (GenPept accession XP_005673517.1), and
Equus caballus (GenPept accession XP_005614077.1) was performed using CLC Sequence Viewer 5.5 (CLC bio A/S,
http://www.clcbio.com).
3. Results
3.1. Clinical features
The family included three affected members (II:1, II:2, and III:1), all males. A male in the third generation was adopted at birth and lost contact with other members of the family. Information on this male member was missing (Fig.
1a). We suggest that the mode of inheritance of the pedigree was X-linked or autosomal.
Fig.1
Pedigree structure and patient phenotypes of the family
(a) Pedigree of a family with Nance-Horan syndrome (NHS). The male identified by a question mark may have been affected, and was adopted out of the family. (b) A photograph of an affected male (III:1) showing a long and narrow face, broad base to the nose, and a thin nasal bridge. (c) Screwdriver-shaped incisors and diastema in individual III:1. The right incisor of the patient was broken in his childhood. (d) Slit-lamp photograph of a carrier female (II:3) showing lens opacities in the posterior Y-suture and cortical coralliform opacity in the left eye. (e, f) Bitemporal retraction in two female carriers (II:3 and I:1)
A detailed description of clinical features is given in Table
1. All affected males had received bilateral lensectomy at an early age to remove congenital nuclear cataracts. All presented with bilateral microcornea, nystagmus, and strabismus. Individual III:1 (Fig.
1b) exhibited typical NHS dental anomalies (screwdriver-shaped left incisor and diastema, and the right incisor of the patient was broken in childhood, as shown in Fig.
1c), which were not noted in the other two affected males. All affected males had the same craniofacial dysmorphisms (a long, narrow face with a broad base to the nose and a thin nasal bridge). Other nonocular anomalies, such as MR, brachymetacarpalia, or cardiovascular abnormalities, were not observed in these three males.
Table 1
Clinical features of the affected males and the female carriers in the NHS family
| Subject |
Eye |
BSCVA |
Ocular feature |
Dental anomaly |
Craniofacial dysmorphism |
| Male patients |
| II:1 |
OD |
20/400 |
Underwent bilateral lensectomy due to congenital nuclear cataract, bilateral microcornea, strabismus, and nystagmus |
Screwdriver-shaped left incisor* and diastema |
Long and narrow face, broad base to nose, and thin nasal bridge |
|
OS |
20/160 |
| II:2 |
OD |
20/400 |
Underwent bilateral lensectomy due to congenital nuclear cataract, bilateral microcornea, strabismus, and nystagmus |
Long and narrow face, broad base to nose, and thin nasal bridge |
|
OS |
20/160 |
| III:1 |
OD |
20/120 |
Underwent bilateral lensectomy due to congenital nuclear cataract, bilateral microcornea, strabismus, and nystagmus |
Long and narrow face, broad base to nose, and thin nasal bridge |
|
OS |
20/120 |
|
| Female carriers |
| I:1 |
OD |
20/60 |
Opacities in the Y-suture and cortex of the lens in both eyes |
|
Bitemporal retraction |
|
OS |
20/40 |
| II:3 |
OD |
20/20 |
Lens opacities in the posterior Y-suture |
|
Bitemporal retraction |
|
OS |
20/30 |
Lens opacities in the posterior Y-suture and cortical coralliform opacity |
BSCVA: best spectacle corrected visual acuity; OD: the right eye; OS: the left eye
*The right incisor of the patient was broken in his childhood
Carrier female II:3 had bilateral fine white dots in the Y-suture of the lenses and a prominent white coralliform cortical opacity in the inferior part of the lens in the left eye (Fig.
1d). Individual I:1 exhibited cortical opacities in both eyes. The two females exhibited bitemporal retraction (Figs.
1e and 1f). No other symptoms related to NHS were observed in the two carrier females.
3.2. Exome sequencing
Using the Agilent SureSelect capture system and the bioinformatics pipeline described in Section 2.5, a mean depth of 84-fold was obtained for this sample, and 95.03% of the target exome region was covered at least 20-fold, indicating that sufficient coverage was achieved for variant calling (Table
2). In total, 103 897 single nucleotide variants (SNVs) and 11 656 small InDels were detected (Table
3). Variants leading to protein coding changes, including missense, nonsense, readthrough, and small InDel variants in the coding region, and potential splice site variants (±10 bp of coding sequence (CDS)), were picked first. Then we used ‘1000 Genome’ data, dbSNP, and the HapMap database to filter out polymorphism variants. Variants with a frequency >0.01 in any of these three databases were filtered out. As a result, we identified 1268 rare variants in the affected male. We first screened these variants for inheritance in an X-linked manner and identified 12 variants on the X chromosome (Table
4). Then, we searched for mutations of known genes involved in hereditary cataracts by visual inspection. The variant c.322G>T in the
NHS gene was identified as a candidate mutation.
Table 2
Sequencing quality statistics of exome capture for the proband sample (individual III:1)
| Parameter |
Value |
| Total target exome region (bp) |
51 391 525 |
| Raw reads |
93 225 420 |
| Raw data yield (Mb) |
8390 |
| Reads mapped to genome |
72 489 626 |
| Reads mapped to target region1
|
55 230 353 (59.24%) |
| Data mapped to target region (Mb) |
4316.82 (51.45%) |
| Capture specificity2 (%) |
76.82 |
| Mean depth of target region (fold) |
84 |
| Mean depth of chromosome X (fold) |
48.24 |
| Mean depth of chromosome Y (fold) |
48.37 |
| Rate of nucleotide mismatch (%) |
0.17 |
| Fraction of target covered (%) |
| ≥4-fold |
99.42 |
| ≥10-fold |
98.32 |
| ≥20-fold |
95.03 |
1Reads mapped to target regions are reads that are within or overlap with the target region
2Capture specificity is defined as the percentage of uniquely mapped reads aligning to the target region
Table 3
Variant statistics of exome capture sequencing for the proband sample (individual III:1) and variant filter statistics
| Parameter |
Number
|
| SNV |
InDel |
| Total |
103 897 |
11 656 |
| Alt_start |
14 |
| Synonymous |
12 519 |
| Missense |
8124 |
| Nonsense |
60 |
| Readthrough |
4 |
| Frameshift |
|
92 |
| Inframe |
|
232 |
| Splice |
2299 |
362 |
| Intron |
65 274 |
8979 |
| 5-UTR |
1928 |
205 |
| 3-UTR |
4136 |
588 |
| Intergenic |
9539 |
1198 |
| Hom |
42 783 |
5148 |
| Het |
61 114 |
6508 |
|
| Total (SNVs+InDels) |
115 553 |
| Variants leading to protein coding change |
11 187 |
| Rare variants leading to protein coding change |
1268 |
| Rare variants in chromosome X |
12 |
| Rare variants in NHS gene |
1 |
SNV: single nucleotide variant; InDel: insertion and deletion; Alt_start: start codon change to an alternative start codon, which can be considered as a start codon coding-synonym; UTR: untranslated regions; Hom: homozygous; Het: heterozygous
Table 4
Rare variants leading to protein coding changes on the chromosome X
| Gene |
Function |
Variant |
Frequency
|
rs ID |
| dbSNP |
1000 Genome |
HapMap |
|
CXorf59
|
Missense |
c.1267C>G |
|
CXorf59
|
Frameshift |
c.1267_1268insTG |
|
EDA2R
|
Missense |
c.323C>T |
0.001 |
|
|
rs186691097 |
|
GPR112
|
Inframe |
c.7966_7968delGAT |
|
IL13RA2
|
Missense |
c.907C>A |
|
MAP3K15
|
Splice |
c.3567-8delT |
|
NHS
|
Nonsense |
c.322G>T |
|
NUDT11
|
Splice |
c.1_−1_13delGCTGCCTCGAGGA |
|
PRDX4
|
Splice |
c.242-10 A>T |
0.001 |
|
|
rs513572 |
|
TEX11
|
Splice |
c.1202-4delT |
|
ZBTB33
|
Inframe |
c.563_564insTGA |
|
ZNF182
|
Missense |
c.282C>A |
0.006 |
0.0065 |
|
rs150066211 |
rs ID: reference SNP ID number
3.3.
NHS mutation analysis and bioinformatics analysis
A previously undescribed nonsense mutation, c.322G>T, in exon 1 of the
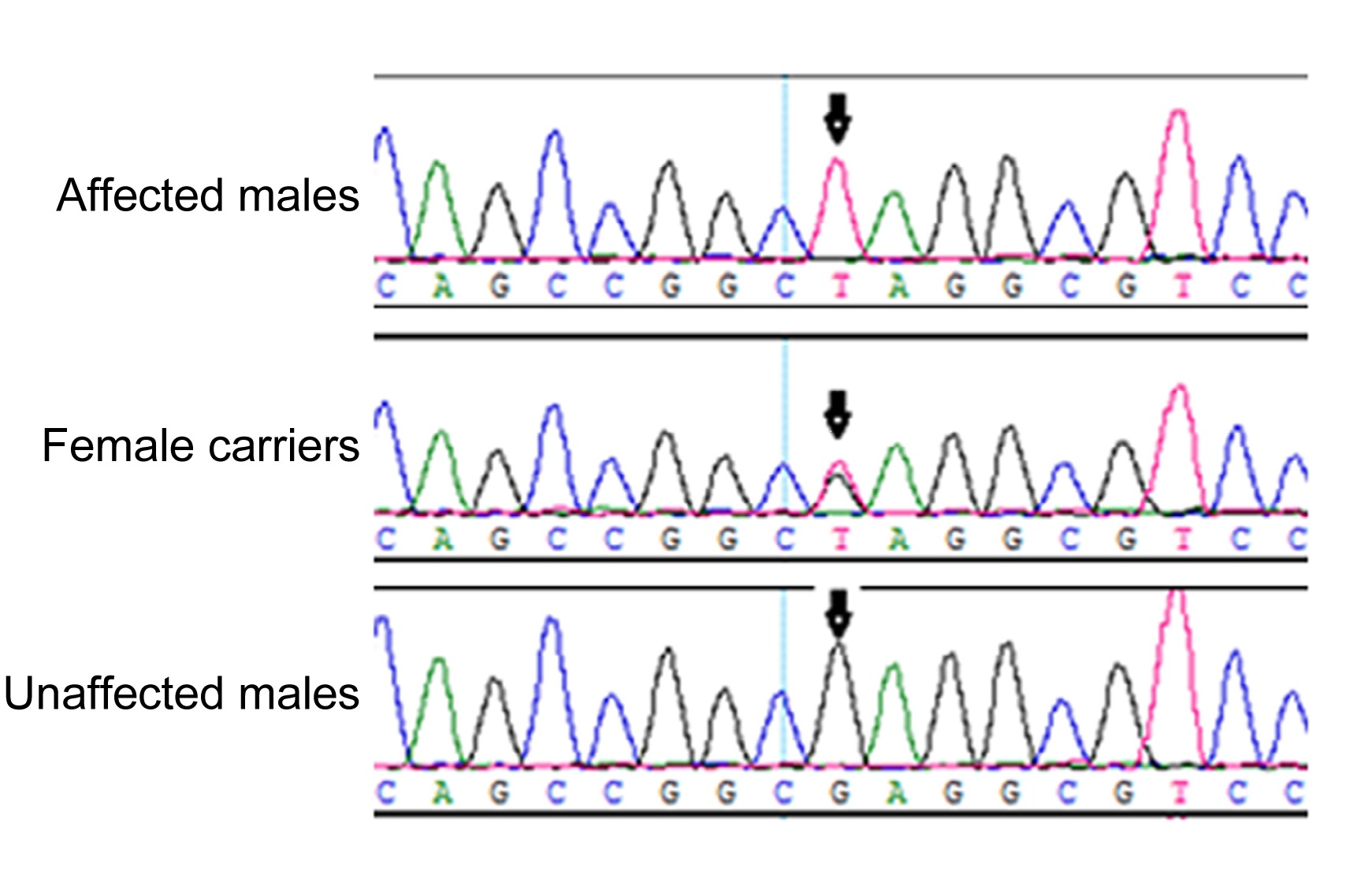
NHS gene (NM_198270), was identified in the three affected males and in the two carrier females (Fig.
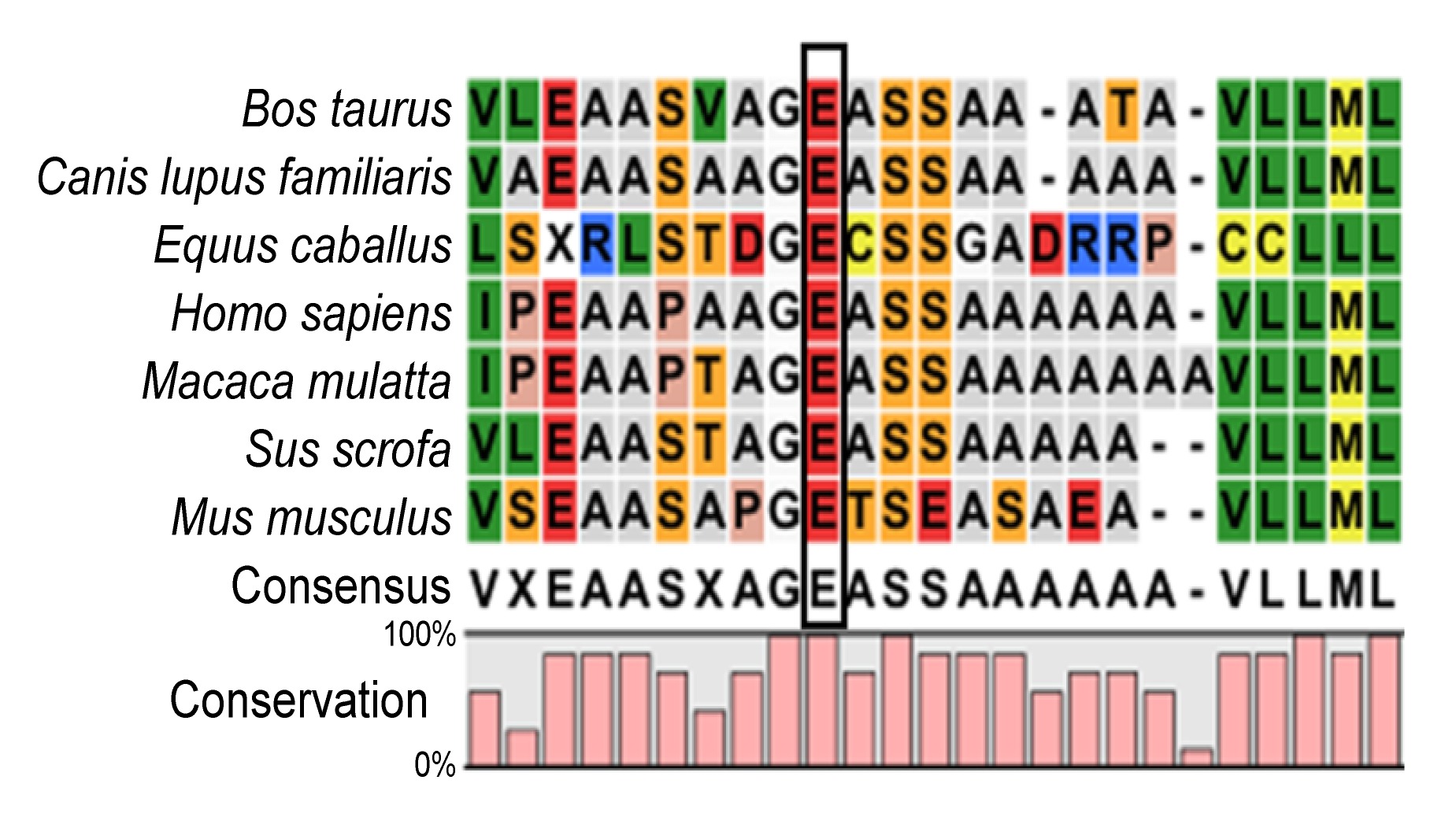
2), but was not detected in the unaffected males or normal controls. This mutation resulted in conversion of glutamic acid to a stop codon at position 108 (E108X). The mutation was not documented in any current database (1000 Genomes Project or HapMap), indicating that the mutation was unlikely to represent an SNP. Multiple sequence alignments were performed and we observed that codon 108, where the mutation (c.322G>T) occurred, was located within a phylogenetically conserved region (Fig.
3).
Fig.2
DNA sequence chromatograms
A nonsense mutation in exon 1, c.322G>T, resulting in E108X in the three affected males and two female carriers, and the normal sequence from two unaffected males
Fig.3
Multiple-sequence alignment of NHS from different species
Multiple-sequence alignment of NHS from different species revealed that codon 108, where the mutation (E108X) occurred, was located within a highly conserved region
4. Discussion
In the present study, we characterized a Chinese family with NHS. Molecular genetic analysis identified a novel nonsense mutation c.322G>T (E108X) in exon 1 of the
NHS gene in all affected males and carrier females. The mutation was not detected in unaffected males or in normal controls. This substitution was conserved in seven tested species, confirming it as a co-segregating disease-causing mutation in the family.
NHS is a rare X-linked disorder, usually underdiagnosed by ophthalmologists with subtle nonocular manifestations that are easily overlooked (Ding et al.,
2009). The affected males in the Chinese family presented with congenital dense cataract, microcornea, nystagmus, and strabismus; nonocular symptoms were not investigated at the initial meeting. We performed exome sequencing of DNA from an affected male (III:1) and identified a mutation in the
NHS gene, which was then shown to segregate with the phenotype in the pedigree. After identification of the mutation, we recalled the family to examine the pedigree members comprehensively. All three affected males manifested craniofacial dysmorphisms characteristic of NHS. Individual III:1 (Fig.
1b) also exhibited typical NHS dental anomalies (screwdriver-shaped incisors and diastema). Among the heterozygous females, carrier signs of NHS were also detected, including lens opacities in the posterior Y-sutures and subtle craniofacial dysmorphism (bitemporal retraction). All of the observations indicated the presence of an NHS phenotype in the family, consistent with the results of the molecular genetic analysis.
NHS is a syndrome with high phenotypic heterogeneity. Tug et al. (
2013) summarized the phenotypes of patients with different mutations and found no obvious genotype-phenotype correlations between the position of each mutation and clinical severity. In our study, nonocular features among the members of the family were also not identical; for example, dental anomalies were observed only in individual III:1 (Fig.
1b). The phenotypic heterogeneity may be due to the role of modifier genes in nonocular tissues (Sharma et al.,
2006).
The exact biological function and structure of the NHS protein remain unclear. On the basis of position-specific iterated BLAST (PSI-BLAST) analysis, Huang et al. (
2006) reported that the N-terminus of the mouse
Nhs1 protein contained a poly-proline domain and a Wiskott-Aldrich syndrome protein (WASP) homology 1 (WH1) domain. Both of these domains are known to be related to targeting proteins to the cytoskeleton. Evidence from a study of Brooks et al. (
2010) showed that the N-termini of isoforms NHS-1A and NHS-A contain a WASP family verprolin-homologous (WAVE) domain (WHD), functionally homologous to the WHD in WAVE proteins. This can interact with members of the Abelson-interactor (Abi) protein family, such as Abi1 and Abi2, playing a regulatory role in actin remodelling and maintaining cell morphology.
In the present study, we describe the first Chinese case of NHS caused by a nonsense mutation in the
NHS gene. The mutation (c.322G>T) induces a putative premature stop codon at nucleotide 322 (E108X), producing a truncated NHS protein. The abnormal protein may initiate the nonsense-mediated mRNA decay (NMD) pathway, a well-known translation-coupled quality control system that degrades mutant mRNA containing premature termination codons (PTCs) (Khajavi et al.,
2006). NMD can protect cells from the toxic effects of truncated proteins caused by dominant-negative or gain-of-function effects (Khajavi et al.,
2006). Previously, Ramprasad et al. (
2005) performed reverse transcription-PCR (RT-PCR) on DNA samples from an Indian NHS family with the c.115C>T (Q39X) mutation in exon 1 of the
NHS gene and found no evidence of NMD.
It has been reported that some mRNAs harbouring PTCs located in some specific regions can avoid the NMD pathway, leading to the translation of abnormal mRNAs (Khajavi et al.,
2006). If the mRNA containing the c.322G>T (E108X) mutation in the present study escaped from the NMD pathway, amounts of truncated protein with possibly destructive effects would be produced in cells. Also, the E108X mutation may damage the function of the WHD of the NHS protein, for example by disrupting the interaction between the WHD and some other functional protein, resulting in an NHS phenotype in patients. However, the pathogenic mechanism of NHS should be investigated in depth in future.
In summary, we identified a novel nonsense mutation (c.322G>T) in exon 1 of the
NHS gene in a Chinese family, which leads to the conversion of glutamic acid to a stop codon (E108X). Our study shows that whole exome analysis in a single affected male from a family exhibiting X-linked inheritance could be used efficiently to identify causative mutations on the chromosome X, thus providing help to clinicians in making a definitive diagnosis of rare diseases.
Acknowledgements
We greatly appreciate the invaluable cooperation and participation of the family members in the present study.
* Project supported by the Science and Technology Specific Project of Zhejiang Province (No. 2009C03010-2), ChinaCompliance with ethics guidelines Nan HONG, Yan-hua CHEN, Chen XIE, Bai-sheng XU, Hui HUANG, Xin LI, Yue-qing YANG, Ying-ping HUANG, Jian-lian DENG, Ming QI, and Yang-shun GU declare that they have no conflict of interest.References
[1] Brooks, S.P., Ebenezer, N.D., Poopalasundaram, S., 2004. Identification of the gene for Nance-Horan syndrome (NHS).
J Med Genet, 41(10):768-771.


[2] Brooks, S.P., Coccia, M., Tang, H.R., 2010. The Nance-Horan syndrome protein encodes a functional wave homology domain (WHD) and is important for co-ordinating actin remodelling and maintaining cell morphology.
Hum Mol Genet, 19(12):2421-2432.
[3] Burdon, K.P., Mckay, J.D., Sale, M.M., 2003. Mutations in a novel gene,
NHS, cause the pleiotropic effects of Nance-Horan syndrome, including severe congenital cataract, dental anomalies, and mental retardation.
Am J Hum Genet, 73(5):1120-1130.
[4] Chograni, M., Rejeb, I., Jemaa, L.B., 2011. The first missense mutation of
NHS gene in a Tunisian family with clinical features of NHS syndrome including cardiac anomaly.
Eur J Hum Genet, 19(8):851-856.
[5] Coccia, M., Brooks, S.P., Webb, T.R., 2009. X-linked cataract and Nance-Horan syndrome are allelic disorders.
Hum Mol Genet, 18(14):2643-2655.
[6] Ding, X., Patel, M., Herzlich, A.A., 2009. Ophthalmic pathology of Nance-Horan syndrome: case report and review of the literature.
Ophthalmic Genet, 30(3):127-135.
[7] Florijn, R.J., Loves, W., Maillette de Buy Wenniger-Prick, L.J., 2006. New mutations in the
NHS gene in Nance-Horan syndrome families from the Netherlands.
Eur J Hum Genet, 14(9):986-990.
[8] Horan, M.B., Billson, F.A., 1974. X-linked cataract and Hutchinsonian teeth.
Aust Paediatr J, 10(2):98-102.
[9] Huang, K.M., Wu, J., Duncan, M.K., 2006. Xcat, a novel mouse model for Nance-Horan syndrome inhibits expression of the cytoplasmic-targeted
Nhs1 isoform.
Hum Mol Genet, 15(2):319-327.
[10] Huang, K.M., Wu, J., Brooks, S.P., 2007. Identification of three novel
NHS mutations in families with Nance-Horan syndrome.
Mol Vis, 13:470-474.
[11] Khajavi, M., Inoue, K., Lupski, J.R., 2006. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease.
Eur J Hum Genet, 14(10):1074-1081.
[12] Khan, A.O., Aldahmesh, M.A., Mohamed, J.Y., 2012. Phenotype-genotype correlation in potential female carriers of X-linked developmental cataract (Nance-Horan syndrome).
Ophthalmic Genet, 33(2):89-95.
[13] Nance, W.E., Warburg, M., Bixler, D., 1974. Congenital X-linked cataract, dental anomalies and brachymetacarpalia.
Birth Defects Orig Artic Ser, 10(4):285-291.
[14] Ramprasad, V.L., Thool, A., Murugan, S., 2005. Truncating mutation in the
NHS gene: phenotypic heterogeneity of Nance-Horan syndrome in an Asian Indian family.
Invest Ophthalmol Vis Sci, 46(1):17-23.
[15] Sharma, S., Ang, S.L., Shaw, M., 2006. Nance-Horan syndrome protein, NHS, associates with epithelial cell junctions.
Hum Mol Genet, 15(12):1972-1983.
[16] Sharma, S., Burdon, K.P., Dave, A., 2008. Novel causative mutations in patients with Nance-Horan syndrome and altered localization of the mutant NHS-A protein isoform.
Mol Vis, 14:1856-1864.
[17] Toutain, A., Ayrault, A.D., Moraine, C., 1997. Mental retardation in Nance-Horan syndrome: clinical and neuropsychological assessment in four families.
Am J Med Genet, 71(3):305-314.
[18] Tug, E., Dilek, N.F., Javadiyan, S., 2013. A Turkish family with Nance-Horan syndrome due to a novel mutation.
Gene, 525(1):141-145.
[19] Walpole, I.R., Hockey, A., Nicoll, A., 1990. The Nance-Horan syndrome.
J Med Genet, 27(10):632-634.

Open peer comments: Debate/Discuss/Question/Opinion
<1>