1. Introduction
Axenfeld-Rieger syndrome (ARS; OMIM 180500) is a common form of anterior segment dysgenesis (ASD) disorder that encompasses a series of clinically and genetically heterogeneous conditions with several developmental abnormalities involving both ocular and systemic structures (Tümer and Bach-Holm,
2009; Chang et al.,
2011; Reis et al.,
2012). Affected individuals display overlapping phenotypes including Axenfeld anomaly, Axenfeld syndrome, Rieger anomaly, and Rieger syndrome (Espinoza et al.,
2002). The classic ocular anterior segment features in ARS include iris hypoplasia, corectopia, polycoria, iridocrneal adhesions, and posterior embryotoxon (a prominent Schwalbe’s line) (Tümer and Bach-Holm,
2009). In addition to the ocular phenotype, systemic anomalies may also be associated with ARS, including midface anomalies, hypertelorism, telecanthus, broad and flat nasal bridge, maxillary hypoplasia with protruding lower lip, dental abnormalities (microdontia, hypodontia, oligodontia, and adontia), and redundant periumblical skin (Tümer and Bach-Holm,
2009). Pituitary abnormalities, hearing defects, congenital cardiac or kidney abnormalities, hypospadia in males, anal stenosis, and growth retardation may also be found (Weisschuh,
2006; Tümer and Bach-Holm,
2009). Because of the severe disruptions of the anterior segment of the eye, ARS patients are at high risk of glaucoma, with approximately 50% of them developing glaucoma throughout their lives (Idrees et al.,
2006; Tümer and Bach-Holm,
2009; D′Haene et al.,
2011).
ARS is a rare autosomal dominant disorder. The incidence of the disease is estimated to be approximately 1:200 000 (Weisschuh,
2006). To date, four genetic loci have been associated with ARS (Weisschuh,
2006), among which two transcription factor-encoding genes,
PITX2 (paired-like homeodomain transcription factor 2; OMIM 601542) at 4q25 and
FOXC1 (forkhead box C1; OMIM 601090) at 6p25, have been identified in ARS patients respectively (Weisschuh,
2006; Kelberman et al.,
2011). Disease-causing mutations and copy number changes in these two genes,
PITX2 and
FOXC1, are estimated to explain approximately 40% of ARS (D′Haene et al.,
2011; Reis et al.,
2012). Two other loci on chromosomes 13q14 and 16q24, supported by linkage analyses, have been suggested to be associated with ARS, but the disease-causing genes have not yet been identified (Tümer and Bach-Holm,
2009; Kelberman et al.,
2011). In addition, the association between ARS and
PAX6 deletion reported by Riise et al. (
2001) has been shown to be incorrect recently (Riise et al.,
2009).
The
PITX2 gene consists of six exons and encodes a bicoid-like homeodomain transcription factor involved in embryogenesis and regulating the development of different tissues of the anterior segment (D′Haene et al.,
2011; Kelberman et al.,
2011). This gene is the first gene to be associated with ARS and produces four alternative transcripts (
PITX2A-
D) (D′Haene et al.,
2011; Reis et al.,
2012). The isoforms (PITX2A, PITX2B, and PITX2C) vary only in their N-terminus, but share the identical 60-amino-acid homeodomain (HD) and C-terminal domain. The common C-terminal domain contains a conserved 14-amino-acid OAR (otp, aristaless, and rax), which is predicted to mediate PITX2 protein interactions (Tümer and Bach-Holm,
2009; Kelberman et al.,
2011). However, a translation product of PITX2D has never been detected (Tümer and Bach-Holm,
2009). PITX2A, the smallest isoform (32 kDa), is the best studied isoform in ARS. The single-exon gene
FOXC1, located on 6p25, is a member of the forkhead (FOX) transcription factor family that is characterized by a conserved 110-amino acid DNA-binding domain and is essential in embryogenesis, tissue-specific gene expression, and tumor development (Tümer and Bach-Holm,
2009; D′Haene et al.,
2011; Kelberman et al.,
2011).
In this study, we presented the clinical features and genetic defects of a Chinese family affected by ARS with several affected individuals presenting variable ocular phenotypes.
2. Materials and methods
2.1. Study subjects and clinical evaluations
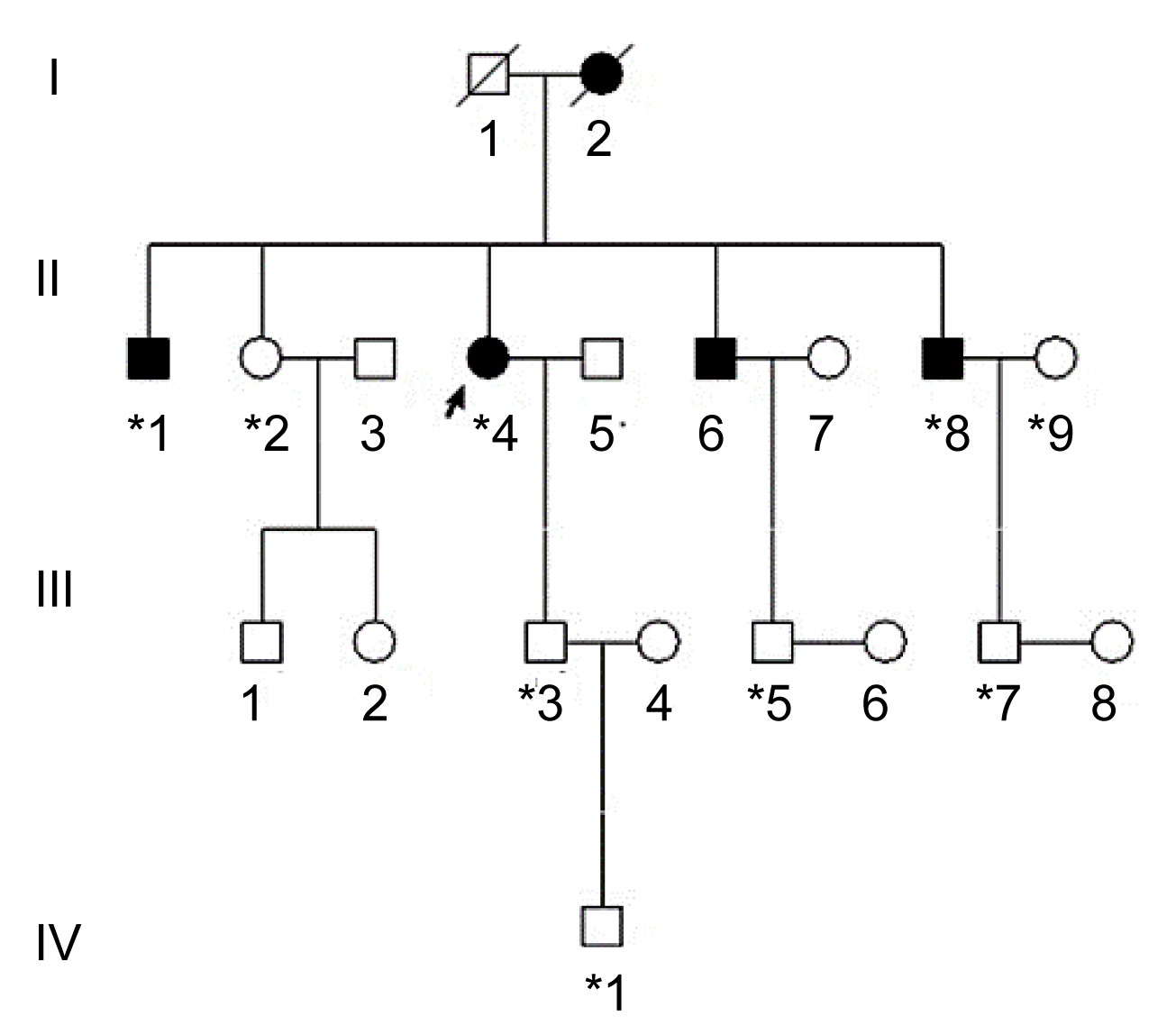
Three generations of a Chinese family with ARS from Zhejiang Province in southeastern China were recruited from the Eye Center of the Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China (Fig.
1). The study was approved by the Institutional Review Board of Zhejiang Institutional Review Board and adhered to the tenets of the Declaration of Helsinki, and informed consent was obtained from all participants. In total, 9 individuals (3 affected and 6 unaffected) from the family participated in the study (Fig.
1). In addition, the medical records of the deceased affected family member (I:2) were reviewed. All participants underwent full ophthalmologic examinations including visual acuity, slit lamp, funduscopy, gonioscopy, and intraocular pressure (IOP) measurements. The phenotypes were documented using slit lamp photography. Additionally, we recruited 100 unrelated ethnically-matched controls with no family history of congenital, heritage ocular diseases.
Fig.1
Pedigree of the family with Axenfeld-Rieger syndrome
*Family members who underwent clinical and genetic analyses. Arrow signals the proband. Squares and circles indicate males and females, respectively. Black and white symbols represent affected and unaffected individuals, respectively
2.2. Mutation analysis
About 2 ml of peripheral blood samples from the family members and the controls involved in the study were drawn by venipuncture and collected in Vacutainer tubes (Becton-Dickinson, Franklin Lakes, NJ) containing EDTA. Genomic DNA was extracted using the Simgen Blood DNA mini kit (Hangzhou Simgen Biotechnology Co., Ltd., Hangzhou, China). Genomic DNA samples from affected and unaffected individuals of the family were used for mutational analysis. All coding exons and intron-exon junctions of
PITX2A and
FOXC1 were amplified using polymerase chain reaction (PCR) with previously published primer sequences (Kelberman et al.,
2011). The PCR cycling conditions were as follows: 95 °C preactivation for 5 min, 10 cycles of touchdown PCR with a 1 °C reduction per cycle from 60 to 50 °C, followed by 35 cycles with denaturation at 95 °C for 25 s, annealing at 55 °C for 25 s, and extension at 72 °C for 1 min, and a final extension at 72 °C for 10 min. PCR products were isolated using electrophoresis on 2% agarose gels and sequenced using the BigDye Terminator Cycle sequencing kit V 3.1 (ABI-Applied Biosystems, Sangon Co., China) on an ABI PRISM 3730 Sequence Analyzer (ABI), according to the manufacturer’s instructions. Sequencing results were analyzed using Chromas 1.62 and compared with sequences from NCBI GenBank (
PITX2A: NM_153427.1 and
FOXC1: NM_001453.2). In addition, PCR products harboring this mutation were further analyzed by clone sequencing, using the method described previously (Zhao et al.,
2012). Direct sequencing was also used to screen the mutation identified in
PITX2 on 100 ethnically-matched controls to exclude the possibility of single nucleotide polymorphism (SNP).
2.3. Modeling of the homeodomain of PITX2A
3D structures of the wild type and the mutant homeodomain of PITX2A were modeled on the basis of the structure of the human Pituitary homeobox 2 chain P (PDB ID: 2l7f) by SWISS-MODEL (Guex and Peitsch,
1997; Schwede et al.,
2003; Arnold et al.,
2006). The structure was determined using nuclear magnetic resonance (NMR) spectroscopy (Doerdelmann et al.,
2012).
3. Results
3.1. Clinical evaluation
The proband (II:4), 54 years old, was referred to the Eye Center. She underwent cataract surgery in her right eye at another hospital six years ago. Then she had an elevated IOP resistant to medication in her right eye for five years. Finally, the right eye was enucleated with ocular prosthesis implanted. Examination of the left eye revealed corneal edema, iris hypoplasia, corectopia, polycoria, peripheral anterior synechia, cataract, and sclerocornea prominent at the temporal limbus. In addition, pigment deposition on nasal and temporal peripheral corneal endothelium was present (Fig.
2a). Funduscopy showed glaucomatous atrophy of the optic nerve with a cup/disc ratio of 0.7 in the left eye, and the IOP was 25 mmHg. Her visual acuity was 20/100 in the left eye. Ocular changes of iris hypoplasia, corectopia, polycoria, peripheral anterior synechia, cataract, and glaucomatous atrophy of the optic nerve in the right eye were also noted in her medical records. Further physical examination revealed characteristic midface anomalies including telecanthus, broad, flat nasal bridge, a thin upper lip, and a protruding lower lip (Figs.
2d and 2e). Dental abnormalities were also present (Fig.
2e). The umbilicus was normal.
Fig.2
Ocular and systemic characteristics of the family with Axenfeld-Rieger syndrome
(a) Left eye of the proband (II:4) showed iris hypoplasia, corectopia, polycoria, corneal edema, sclerocornea prominent at the temporal limbus, and pigment deposition on nasal and temporal peripheral corneal endothelium; (b, c) Right and left eyes, respectively, of the brother of the proband (II:8) showed mild corectopia and iris hypoplasia; (d) Midface abnormalities of the proband (II:4) included telecanthus, broad and flat nasal bridge (Note that the right eye was prosthetic eye); (e) Systemic abnormalities of the proband (II:4) also included a thin upper lip, a protruding lower lip and dental abnormalities (Note that the maxillary teeth of the proband were dental prosthesis); (f) The absence of the maxillary anterior teeth was observed from the brother of the proband (II:8)
Individual II:1, 62 years old, was one of the proband’s brothers with bilateral ocular globe atrophy after severe ocular trauma when he was three years old. He had no light perception. Although there was no record of the ocular appearance, he had the similar nonocular anomalies as the proband. Individual II:8 was another brother of the proband. Mild corectopia and iris hypoplasia were detected in ocular examination. Both the fundus and IOP were normal. He also had the similar nonocular anomalies as the proband (Fig.
2f). His visual acuity was 20/25 in both eyes. Individual II:2, the sister of the proband, had no similar ocular and nonocular anomalies. Other family members including individual III:2, IV:1, III:5, and III:7 were examined, and no similar anomaly was noted. Individual I:2, the mother of the proband, had signs of ARS including iris hypoplasia, corectopia, polycoria, peripheral anterior synechia, midface anomalies, and dental anomalies according to the medical records. The clinical evaluation of the affected individuals is summarized in Table
1.
Table 1
Clinical details of affected individuals
| Patient |
Age (year) |
Visual acuity |
IOP (mmHg) |
Ocular feature |
Extraocular feature |
| I:2*
|
NA |
NA |
NA |
OU: iris hypoplasia, corectopia, polycoria, peripheral anterior synechia |
Midface abnormalities, dental abnormalities |
| II:1 |
62 |
No light perception |
NA |
OU: ocular globe atrophy |
Midface abnormalities, dental abnormalities |
| II:4 |
54 |
20/100 (OS) |
25 |
OD: prosthetic eye; OS: iris hypoplasia, corectopia, polycoria, peripheral anterior synechia, cataract, corneal edema, sclerocornea, sclerocornea, pigment deposition on corneal endothelium, optic nerve atrophy |
Midface abnormalities, dental abnormalities |
| II:6 |
50 |
NA |
NA |
NA |
Midface abnormalities, dental abnormalities |
| II:8 |
48 |
20/25 (OD); 20/25 (OS) |
17 (OD); 18 (OS) |
OU: mild corectopia and iris hypoplasia |
Midface abnormalities, dental abnormalities |
*The data were according to the medical records of the patient before her death
OD: right eye; OS: left eye; OU: both eyes; IOP: intraocular pressure; NA: not available
3.2. Clinical evaluation
Direct sequencing of all coding exons and intron-exon junctions of the
PITX2 gene revealed a heterozygous variation involving multiple nucleotides in exon 5 in all affected individuals (Fig.
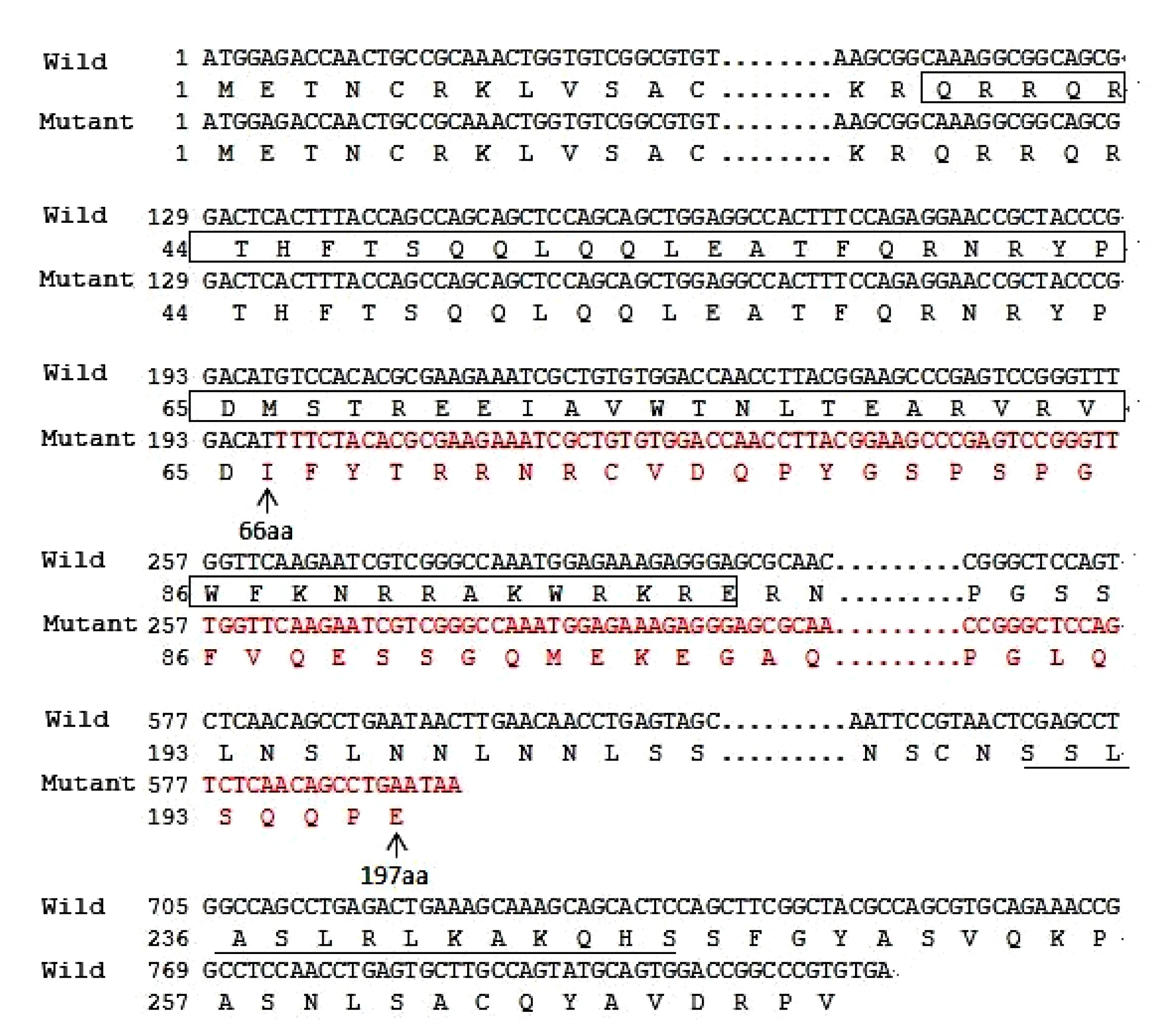
3c). After cloning sequencing, we found a deletion/insertion mutation in the mutated allele, c.198_201delinsTTTCT (p.M66Ifs*133) in exon 5 (Fig.
3b). It resulted in a frameshift in codon 66 and was supposed to produce a truncated protein with only 197 amino acids including an aberrant 132 residues relative to the wild type PITX2A protein (Fig.
4). Direct sequencing and cloning sequencing analyses of the members of the family showed that the mutation co-segregated with disease. The mutation was not present in unaffected family members or 100 normal control individuals. No sequence alterations were detected in the
FOXC1 gene. This suggests that this novel complex deletion/insertion mutation in
PITX2 is a disease-causing mutation for ARS in this family.
Fig.3
Identification of the novel c.198_201delinsTTTCT mutation in the studied family
(a) Sequences from a normal individual; (b) Cloning sequencing showing the deletion/insertion mutation c.198_201delinsTTTCT; (c) Sequence of an affected individual with heterozygous mutation c.198_201delinsTTTCT
Fig.4
Amino acid sequences for the wild type and mutant PITX2A protein
c.198_201delinsTTTCT causes frameshift and substitutes amino acids 66–197 by abnormal residues (red), and results in a truncated PITX2A protein with disrupted homeodomain (box). In addition, it lacks the 14-amino acid OAR domain (underlined) in the C-terminus (Note: for interpretation of the references to color in this figure legend, the reader is referred to the web version of this article)
3.3. Comparison of wild type and mutant structures of the homeodomain of PITX2A
The p.M66Ifs*133 mutation was supposed to produce a premature stop codon at codon 198 in exon 6. A truncated protein with 197 amino acids including an aberrant 132-amino acid residue was putatively generated, 74 amino acids less than the wild type PITX2A protein, which possessed 271 amino acids (Fig.
4). When analyzed by SWISS-MODEL, only the first helical region (helix 1) was conserved, and the other two helical regions (helixes 2 and 3) were disrupted (Fig.
5).
Fig.5
Comparison of wild type (a) and mutant (b) structures of the homeodomain of PITX2A by SWISS-MODEL
The wild type structural model of the homeodomain of PITX2A contains three helical regions (helixes 1, 2, and 3). The mutant structural shows that helix 1 is conserved, but helixes 2 and 3 are disrupted
4. Discussion
In this study, we identified a novel deletion/insertion mutation (c.198_201delinsTTTCT) in the
PITX2 gene in a Chinese ARS family. The mutation co-segregated with anterior segment defects and systemic anomalies in this family. ARS caused by the same
PITX2 mutation exhibited considerable phenotypic heterogeneity among affected individuals in a single pedigree in this study, which is consistent with previous studies (Li et al.,
2008; Tümer and Bach-Holm,
2009; Kelberman et al.,
2011; Law et al.,
2011). The brother of the proband had mild corectopia and iris hypoplasia, whereas the proband showed almost the full spectrum of the anterior segment defects except posterior embryotoxon. Although there is no clear genotype-phenotype relationship,
PITX2 mutations are usually related to ARS patients with systemic changes such as dental and umbilical anomalies, whereas mutations in
FOXC1 appear to be associated with isolated ocular or ocular, heart, and/or hearing defects, such as congenital mitral valve abnormality and sensorineural hearing loss (Strungaru et al.,
2007; Tümer and Bach-Holm,
2009; D′Haene et al.,
2011; Reis et al.,
2012; Gripp et al.,
2013).
PITX2 mutation in this study was also associated with systemic changes including midface and dental anomalies.
PITX2 is a bicoid-related homeodomain transcription factor required for the development and maintenance of the ocular anterior segment tissues and nonocular tissues including the pituitary, heart, and branchial arches (Gage et al.,
1999; Kitamura et al.,
1999; Kelberman et al.,
2011; Doerdelmann et al.,
2012). PITX2 is dosage-sensitive and either increased or decreased alteration causes developmental disorders, such as ARS and glaucoma (Tümer and Bach-Holm,
2009). PITX2A, the smallest isoform (32 kDa) and the best studied isoform in ARS, consists of five regions: two activation domains (AD1 and AD2), one homeodomain (HD), and two inhibitory domains (ID1 and ID2) (Footz et al.,
2009; D′Haene et al.,
2011). AD1 is located at the N-terminus, ID1, AD2, and ID2 are located at the C-terminus of the PITX2A. The 60-amino acid homeodomain consists of three helices in a compactly folded structure and a flexible N-terminal arm, responsible for the recognition and binding to target DNA sequences (Doerdelmann et al.,
2012). Helix 3, also called the recognition helix, inserts itself into the major groove of the recognition site and establishes contacts with the specific residues of the bicoid binding sequence (Li et al.,
2008; Doerdelmann et al.,
2012). Helix 2 makes DNA backbone contacts, enhancing specific homeodomain-DNA interactions. In addition to the helices, the N-terminal arm makes contacts with the bases in the DNA minor groove (Doerdelmann et al.,
2012).
To our knowledge, more than 50 intragenic
PITX2 mutations have been described in Table
S1. However, only one deletion/insertion mutation has been reported previously (Perveen et al., 2000). Among all these mutations, frameshift, splice-site and nonsense mutations are scattered along the coding region and may result in truncated proteins, whereas most of the missense mutations are within the homeodomain (Footz et al., 2009). In vitro assays are often used to investigate the effects of the mutant PITX2 proteins, including subcellular distribution, DNA binding, transactivation activity, and protein stability (Footz et al., 2009; Quentien et al., 2010). In general, as nonsense and frameshift mutations result in truncated and therefore nonfunctional proteins, they are considered to be pathogenic (D′Haene et al., 2011). To date, several frameshift mutations within the homeodomain of PITX2 have been identified, and these mutations lead to severe ocular phenotypes (Priston et al., 2001; Wang et al., 2003; Tümer and Bach-Holm, 2009; Reis et al., 2012). For example, a 21-bp in-frame duplication introduces seven extra amino acids at the junction of the N-terminal arm and helix 1 of the homeodomain, and results in a defective PITX2 protein with reduced DNA-binding activity and no detectable transactivation (Priston et al., 2001). Wang et al. (2003) identified c.134_137delACTT mutation in PITX2 gene, which is predicted to cause a frameshift after Thr44, the 7th amino acid of the homeodomain, and delete all the functionally important parts of the PITX2 protein.
The c.198_201delinsTTTCT (p.M66Ifs*133) mutation identified in the present study is a novel deletion/insertion mutation in exon 5, which causes frameshift after Asp65 of PITX2A, and introduces a premature stop codon in exon 6 (Fig. 4). As this premature stop codon is located in the final exon of the gene, it is not expected to be subject to nonsense-mediated decay (NMD) (Nagy and Maquat, 1998; Wen and Brogna, 2008; Yepiskoposyan et al., 2011; Reis et al., 2012). Intriguingly, this mutation is similar to mutation c.143_144delGC (p.S48Tfs*150) reported by Reis et al. (2012). They both result in a truncated 197-amino acid long protein and most of the frame-shifted amino acids are the same. This frameshift mutation changes the coding sequence from the 28th amino acid residue of the homeodomain, which disrupts helixes 2 and 3 in the homeodomain and the C-terminus of PITX2A (Figs. 4 and 5). Helix 2 and helix 3, the crucial helical regions of the homeodomain, are essential for binding DNA and are critical for the transcription factor function (Dave et al., 2000; Doerdelmann et al., 2012). In addition, the OAR domain in the C-terminal end has been proposed to increase the DNA binding and transcriptional activity of PITX2 during development (Amendt et al., 2000). Thus, this is a loss-of-function mutation, deleting almost all the functionally important parts of the PITX2 protein.
5. Conclusions
In summary, we identified a novel mutation c.198_201delinsTTTCT (p.M66Ifs*133) in a Chinese family affected by ARS. This deletion/insertion mutation causes a frameshift in the homeodomain of the PITX2 protein. Our results further support the critical role of the homeodomain in PITX2 protein and may contribute to the understanding of the pathogenesis causes of ARS.
Acknowledgements
We are grateful to all the family members and the normal volunteers for their participation in the study.
* Project supported by the Qianjiang Talents Project of Zhejiang Province (No. 2010R10067), the Zhejiang Key Innovation Team Project of China (No. 2009R50039), and the Zhejiang Key Laboratory Foundation of China (No. 2011E10006)# Electronic supplementary materials: The online version of this article (http://dx.doi.org/10.1631/jzus.B1300053) contains supplementary materials, which are available to authorized usersCompliance with ethics guidelines Hou-fa YIN, Xiao-yun FANG, Chong-fei JIN, Jin-fu YIN, Jin-yu LI, Su-juan ZHAO, Qi MIAO, and Feng-wei SONG declare that they have no conflict of interest.
References
[1] Amendt, B.A., Semina, E.V., Alward, W.L., 2000. Rieger syndrome: a clinical, molecular, and biochemical analysis.
Cell Mol Life Sci, 57(11):1652-1666.


[2] Arnold, K., Bordoli, L., Kopp, J., 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling.
Bioinformatics, 22(2):195-201.
[3] Chang, T.C., Summers, C.G., Schimmenti, L.A., 2011. Axenfeld-Rieger syndrome: new perspectives.
Br J Ophthalmol, 96(3):318-322.
[4] Dave, V., Zhao, C., Yang, F., 2000. Reprogrammable recognition codes in bicoid homeodomain-DNA interaction.
Mol Cell Biol, 20(20):7673-7684.
[5] DHaene, B., Meire, F., Claerhout, I., 2011. Expanding the spectrum of
FOXC1 and
PITX2 mutations and copy number changes in patients with anterior segment malformations.
Invest Ophthalmol Vis Sci, 52(1):324-333.
[6] Doerdelmann, T., Kojetin, D.J., Baird-Titus, J.M., 2012. Structural and biophysical insights into the ligand-free Pitx2 homeodomain and a ring dermoid of the cornea inducing homeodomain mutant.
Biochemistry, 51(2):665-676.
[7] Espinoza, H.M., Cox, C.J., Semina, E.V., 2002. A molecular basis for differential developmental anomalies in Axenfeld-Rieger syndrome.
Hum Mol Genet, 11(7):743-753.
[8] Footz, T., Idrees, F., Acharya, M., 2009. Analysis of mutations of the
PITX2 transcription factor found in patients with Axenfeld-Rieger syndrome.
Invest Ophthalmol Vis Sci, 50(6):2599-2606.
[9] Gage, P.J., Suh, H., Camper, S.A., 1999. Dosage requirement of
PITX2 for development of multiple organs.
Development, 126(20):4643-4651.
[10] Gripp, K.W., Hopkins, E., Jenny, K., 2013. Cardiac anomalies in Axenfeld-Rieger syndrome due to a novel
FOXC1 mutation.
Am J Med Genet Part A, 161(1):114-119.
[11] Guex, N., Peitsch, M.C., 1997. SWISS-MODEL and the SWISS-Pdb Viewer: an environment for comparative protein modeling.
Electrophoresis, 18(15):2714-2723.
[12] Idrees, F., Vaideanu, D., Fraser, S.G., 2006. A review of anterior segment dysgeneses.
Survey Ophthalmol, 51(3):213-231.
[13] Kelberman, D., Islam, L., Holder, S.E., 2011. Digenic inheritance of mutations in
FOXC1 and
PITX2: correlating transcription factor function and Axenfeld-Rieger disease severity.
Hum Mutat, 32(10):1144-1152.
[14] Kitamura, K., Miura, H., Miyagawa-Tomita, S., 1999. Mouse
PITX2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism.
Development, 126(24):5749-5758.
[15] Law, S.K., Sami, M., Piri, N., 2011. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel
PITX2 mutation.
Mol Vis, 17:1231-1238.
[16] Li, D., Zhu, Q., Lin, H., 2008. A novel
PITX2 mutation in a Chinese family with Axenfeld-Rieger syndrome.
Mol Vis, 14:2205-2210.
[17] Nagy, E., Maquat, L.E., 1998. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance.
Trends Biochem Sci, 23(6):198-199.
[18] Perveen, R., Lloyd, I.C., Clayton-Smith, J., 2000. Phenotypic variability and asymmetry of Rieger syndrome associated with
PITX2 mutations.
Invest Ophthalmol Vis Sci, 41(9):2456-2460.
[19] Priston, M., Kozlowski, K., Gill, D., 2001. Functional analyses of two newly identified
PITX2 mutants reveal a novel molecular mechanism for Axenfeld-Rieger syndrome.
Hum Mol Genet, 10(16):1631-1638.
[20] Quentien, M.H., Vieira, V., Menasche, M., 2010. Truncation of
PITX2 differentially affects its activity on physiological targets.
J Mol Endocrinol, 46(1):9-19.
[21] Reis, L.M., Tyler, R.C., Volkmann Kloss, B.A., 2012.
PITX2 and
FOXC1 spectrum of mutations in ocular syndromes.
Eur J Hum Genet, 20(12):1224-1233.
[22] Riise, R., Storhaug, K., Brondum-Nielsen, K., 2001. Rieger syndrome is associated with
PAX6 deletion.
Acta Ophthalmol Scand, 79(2):201-203.
[23] Riise, R., DHaene, B., de Baere, E., 2009. Rieger syndrome is not associated with
PAX6 deletion: a correction to
Acta Ophthalmol Scand 2001: 79: 201-203.
Acta Ophthalmol, 87(8):923
[24] Schwede, T., Kopp, J., Guex, N., 2003. SWISS-MODEL: an automated protein homology-modeling server.
Nucl Acids Res, 31(13):3381-3385.
[25] Strungaru, M.H., Dinu, I., Walter, M.A., 2007. Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with
FOXC1 and
PITX2 mutations.
Invest Ophthalmol Vis Sci, 48(1):228-237.
[26] Tmer, Z., Bach-Holm, D., 2009. Axenfeld-Rieger syndrome and spectrum of
PITX2 and
FOXC1 mutations.
Eur J Hum Genet, 17(12):1527-1539.
[27] Wang, Y., Zhao, H., Zhang, X., 2003. Novel identification of a four-base-pair deletion mutation in
PITX2 in a Rieger syndrome family.
J Dent Res, 82(12):1008-1012.
[28] Weisschuh, N., 2006. Novel mutations of
FOXC1 and
PITX2 in patients with Axenfeld-Rieger malformations.
Invest Ophthalmol Vis Sci, 47(9):3846-3852.
[29] Wen, J., Brogna, S., 2008. Nonsense-mediated MRNA decay.
Biochem Soc Trans, 36(Pt 3):514-516.
[30] Yepiskoposyan, H., Aeschimann, F., Nilsson, D., 2011. Autoregulation of the nonsense-mediated mRNA decay pathway in human cells.
RNA, 17(12):2108-2118.
[31] Zhao, Y.E., Wang, Z.H., Xu, Y., 2012. Cloning and sequence analysis of chitin synthase gene fragments of
Demodex mites.
J Zhejiang Univ-Sci B (Biomed & Biotechnol), 13(10):763-768.



Open peer comments: Debate/Discuss/Question/Opinion
<1>