1. Introduction
Acute lung injury (ALI) is one of the most common complications following extracorporeal circulation (ECC), and the mortality rate in its severe form, acute respiratory distress syndrome (ARDS), is very high (Ng et al.,
2002; Staton et al.,
2005). ALI associated with ECC, characterized with pulmonary inflammation, is primarily believed to be due to systemic inflammatory response syndrome (SIRS) (Zhou et al.,
2008;
2010; Tao et al.,
2009). After activated by plasma proinflammatory mediators, neutrophils are recruited in alveolar spaces due to chemokines (Du et al.,
2012; Li et al.,
2012), and further release proteolytic enzymes and free radicals, which can cause damages to pulmonary endothelial cells as well as epithelial cells, leading to increased pulmonary alveolar-endothelial permeability and affecting gas exchange (Zhou et al.,
2008; Tao et al.,
2009). Therefore, inhibiting the accumulation of neutrophils in the lung may mitigate ECC-associated ALI (Zhou et al.,
2010; Du et al.,
2012).
Tumor necrosis factor-α (TNF-α) is a highly potent proinflammatory cytokine, which is primarily produced by activated macrophages and monocytes (Roach et al.,
2002; Du et al.,
2012). The biological functions of TNF-α are involved in the innate immunity, and have a crucial role in pathogenesis of many diseases, such as sepsis. Activation of pulmonary macrophages and release of TNF-α are the earliest manifestations of ALI, which leads to up-regulation of cell surface adhesion molecules and subsequent production of cytokines and chemokines (such as interleukin-6 (IL-6) and IL-8) that induce recruitment of neutrophils within the pulmonary microvasculature (Sedgwick et al.,
2000; Mukhopadhyay et al.,
2006). Overexpression of TNF-α is associated with the increased permeability of pulmonary vasculature (Khimenko et al.,
1999). Kotani et al. (
2000) have shown that TNF-α mRNA level increases 4–6 folds at the end of ECC. Yende et al. (
2003) demonstrated that polymorphisms in the TNF gene (TNF-308) are associated with prolonged mechanical ventilation after a coronary artery bypass graft. Carney et al. (
1999) further demonstrated that TNF receptor-binding protein (TNF-bp) significantly reduced plasma TNF levels and reduced the incidence of ARDS following ECC+lipopolysaccharide (LPS) treatment. Recently, we found that the plasma TNF-α level in ARDS patients was significantly higher than that in non-ARDS patients (Li et al.,
2012). These research studies indicated that TNF-α plays an important role in ECC-induced ALI.
TNF-α has multiple receptors, including TNF receptor 1 (TNFR1), TNFR2, Fas (differentiation 95 (CD95) or apoptosis antigen 1 (Apo-1)), and CD40 (Drost and MacNee,
2002). Previous studies showed that TNFR1 is responsible for neutrophil apoptosis, down-regulates chemokine (C-X-C motif) receptor 2 (CXCR2) expression in neutrophils, and reduces neutrophil infiltration and migration to the infectious site (Secher et al.,
2009). The evidence suggests that TNFR1 pathway plays an important role in inflammatory response associated with neutrophils. In our present study, using an ECC model, we evaluated the role of TNF-TNFR1 pathway in ECC-induced ALI.
2. Materials and methods
2.1. ALI model induced by ECC
ECC was established as reported by Du et al. (
2012). Briefly, male Sprague-Dawley (SD) rats (around 250 g) were anesthetized with pentobarbital (40 mg/kg i.p.), and then ventilated with 40% oxygen. After heparinization (3 mg/kg), the right carotid artery and left femoral vein were cannulated and connected with a roller pump (Stöck II, Munich, Germany) and a tube (1/16 inch internal diameter) primed with Hydroxyethyl Starch 130/0.4. Artery pressure, electrocardiography (ECG), and heart rate were monitored throughout the experiment. All animals were provided by the Animal Experiment Center of Sichuan University, and the protocol was approved by the Animal Hospital Ethics Committee of West China Hospital, Sichuan University, China. Handling of the animals was in compliance with the Guide for the Care and Use of Laboratory Animals.
2.2. Study design
In the present study, CAY10500 (Sigma-Aldrich, St. Louis, MO, USA) was used to block the TNF-TNFR1 pathway. CAY10500 is a small-molecule inhibitor of TNF-α, which promotes subunit disassembly of this trimeric cytokine family member to form a dimeric complex, which cannot bind to TNFR1 and thereby inhibit the downstream signaling pathway, for instance, the activation of nuclear factor-кB (NF-кB) (He et al.,
2005).
To test the effect of CAY10500 on TNFR1, neutrophils adhered to vascular endothelial cells induced by TNF-α were detected
in vitro according to our previous study (Du et al.,
2012; Li et al.,
2012). Briefly, blood was collected from a rat vein, and neutrophils were separated by centrifugation with a neutrophil separating medium at 700×
g for 30 min at 4 °C. After confirmed by Trypan blue staining, neutrophil yields were incubated in Dulbecco’s modified Eagle’s medium (DMEM) at 37 °C for 2 h for resting cells.
Vascular endothelial cells (1×10
5) were grown in 6-well gelatin-coated plates for 48–72 h, and then the harvested neutrophils were added and stimulated with TNF-α (500 pg/ml; from Peprotech, Rocky Hill, NJ, USA) or TNF-α+CAY10500 (0.075 μmol/L) or placebo for 2 h at 37 °C. The cells were washed three times with PBS, and then assayed by a phase contrast microscopy (Nikon, Melville, NY, USA).
To observe the effect of CAY10500 on pulmonary inflammation, 30 animals were randomly assigned to three groups (
n=10 in each group) after establishment of ECC. Rats were pretreated with 0.3 ml Ringer’s solution IV in ECC control group, or 0.3 ml ethanol IV in the vehicle group, or 0.3 mg CAY10500 (diluted in 0.3 ml ethanol) in the CAY group. All animals were on ECC for 2 h and monitored for 4 h after ECC and before the sample collection. In addition, 10 animals on ECC for 1 min were sacrificed to obtain their baseline values.
2.3. Measurements
2.3.1. Oxygenation index (OI)
Artery blood samples were drawn from the right carotid artery. Artery oxygen partial pressure (PaO
2) was measured by ABL800 FLEX (Denmark Radiometer Medical ApS). Then OI was calculated by the formula of PaO
2/FiO
2 (fraction of inspired oxygen).
2.3.2. Bronchoalveolar lavage fluid (BALF) and cell counts
Bronchoalveolar lavage (BAL) was performed based on Du et al. (
2012) at 4 h after ECC. Rats were sacrificed by peritoneal pentobarbital injection. The trachea was cannulated and secured by ligation. Three milliliters of phosphate buffered saline (PBS) were instilled and slowly withdrawn. The collected BALF was centrifuged at 300 r/min for 10 min at 4 °C. Supernatant was stored at −70 °C for future analysis. Precipitate was washed with PBS and resuspended, and then cells were analyzed by an automatic blood cell analyzer (MINDRAY, BC-3000, China).
2.3.3. Determination of TNF-α levels
A blood sample was drawn from the femoral artery and centrifuged at 4 °C. Then 0.5 ml plasma was collected and frozen at −70 °C until later analysis. TNF-α levels in both plasma and BALF were determined by an enzyme-linked immunosorbent assay (ELISA) kit (eBioscience, San Diego, CA, USA) following the manufacturer’s instructions.
2.3.4. Histopathological examination of lungs
Tissues were prepared for light microscopy using standard procedures. The histopathological examination of lungs was performed as we reported before (Tao et al.,
2009; Zhou et al.,
2010; Du et al.,
2012). Briefly, half of the right lungs were rinsed with ice-cold saline. Lung specimens were fixed in 10% formalin, dehydrated, embedded in paraffin, sectioned (3–5 μm), and then stained with hematoxylin-eosin (H&E). Specimens were examined under a light microscope. Neutrophils were calculated by a blinded observer according to the counts in 30 continuous areas 100 μm from the edge.
2.3.5. Measurement of lung wet/dry weight (WW/DW) ratio
The rest of the right lungs of the animals were excised and weighed, and then placed in an oven at 60 °C for 24 h to obtain their DW. The tissue edema was evaluated through calculation of the WW/DW ratio of the lung tissue.
2.4. Statistical analysis
Data were expressed as mean±standard deviation (SD), and the difference between the groups was analyzed by one-way analysis of variance (ANOVA) using SPSS 16.0.
P values of less than 0.05 were considered significant.
3. Results
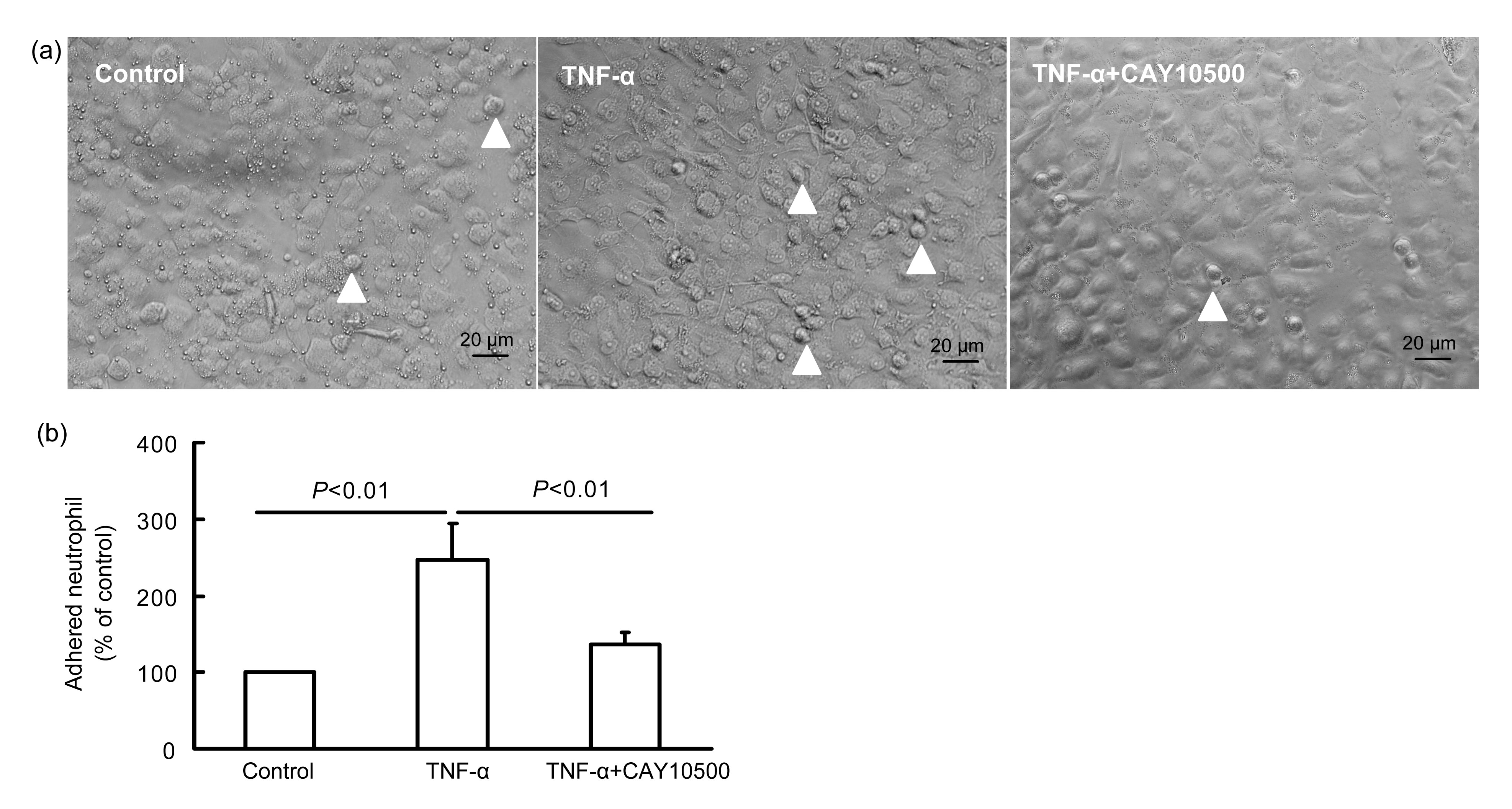
3.1. Effect of CAY10500 on neutrophil adhesion to endothelial cells
Compared with the control, neutrophils adhered to endothelial cells were increased to 2.5-fold under the stimulation of TNF-α (
n=6,
P<0.01; Fig.
1), and the adhesive neutrophils were reduced under the treatment of CAY10500 and TNF-α (
P<0.01 vs. only TNF-α treatment), suggesting that TNFR1 plays an important role in the adhesion of neutrophils and endothelial cells induced by TNF-α.
Fig.1
Effect of CAY10500 on neutrophil adhesion to endothelial cells
The results show the endothelial tissue (a) and neutrophil counts (b) in the experiment. Neutrophil-endothelial cell adhesion (white arrowheads) induced by TNF-α was inhibited by CAY10500 in vitro. Blood was collected from the rat vein, and neutrophils were separated by centrifugation. Vascular endothelial cells were grown in 6-well gelatin-coated plates. Neutrophil and endothelial cells were stimulated with TNF-α (100 μmol/L), or TNF-α+CAY10500 (0.075 μmol/L), or placebo for 2 h at 37 °C. The cells were washed three times with PBS, and then assayed by a phase contrast microscopy
3.2. Effect of CAY10500 on pulmonary function
Pulmonary function was assessed by OI (Fig.
2). Compared with the baseline ((438±59) mmHg), OI values were significantly decreased after ECC in both the ECC control group ((285±44) mmHg) and vehicle group ((240±49) mmHg). And they were slightly improved by CAY10500 pretreatment ((318±72) mmHg,
P>0.05 vs. the control and vehicle groups).
Fig.2
Effect of CAY10500 on pulmonary function
Oxygenation index (OI, PaO2/FiO2) (a) and blood cell counts (b) were not changed by CAY10500 pretreatment. After establishment of the carotid artery-femoral vein ECC model, rats were treat with PBS (ECC control), or 0.3 ml ethanol (vehicle), or 0.3 mg CAY10500 (CAY). All animals were on ECC for 2 h and monitored for 4 h after ECC, and then PaO2 and blood cell counts were measured. In addition, 10 animals on ECC for 1 min were sacrificed to obtain the baseline values
3.3. TNF-α levels in plasma and BALF
Compared with baseline levels ((15.6±5.7) pg/ml), plasma TNF-α levels in the control group increased 10-fold to (157.7±35.0) pg/ml after ECC. Pretreatment of CAY10500 significantly decreased plasma TNF-α levels to (75.5±20.4) pg/ml (
P<0.05 vs. the ECC control group; Fig.
3a).
Fig.3TNF-α levels in plasma (a) and BALF (b) in experimental groups
Rats were treated as described in Fig.
1. The plasma and BALF were harvested after ECC to measure TNF-α. Pretreatment of CAY10500 significantly decreased plasma TNF-α levels to (75.5±20.4) pg/ml and did not obviously reduce the amount of TNF-α in the BALF ((514±140) pg/ml)
TNF-α levels in BALF had a similar trend, but significantly higher than those in plasma (
P<0.01; Fig.
3), indicating that the lung may be the main source of TNF-α during ECC. Compared with the baseline level ((109±39) pg/ml), the BALF TNF-α in the control group greatly increased ((642±76) pg/ml,
P<0.01) after ECC. Pretreatment of CAY10500 slightly reduced BALF TNF-α ((514±140) pg/ml), but without significant differences (
P>0.05; Fig.
3b).
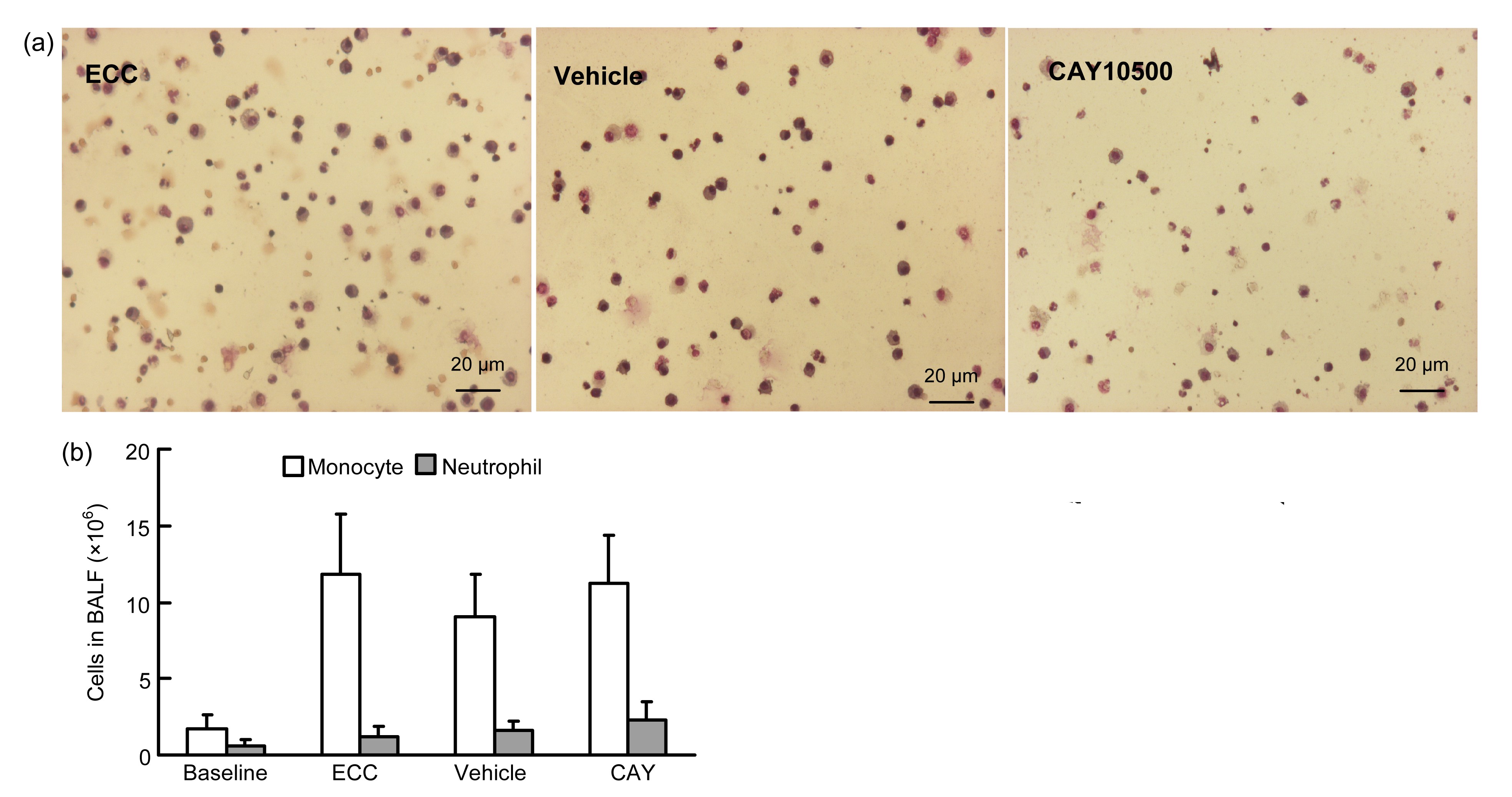
3.4. Neutrophil counts in BALF
Counts of blood leukocytes and neutrophils increased after ECC, but there was no difference between all groups (Fig.
4).
Fig.4Cell counts in BALF
Rats were treated as described in Fig.
2. The BALF was harvested after ECC to measure cell counts. (a) Leukocytes counts/high-power field (HPF); (b) Amount of monocytes and neutrophils in BALF
The amount of neutrophils in BALF was used to assess leukocyte infiltration and ALI. As shown in Fig.
4b, leukocyte counts in BALF significantly increased after ECC from (1.7±0.9)×10
6 to (11.80±6.91)×10
6 and the neutrophil amount greatly increased from (0.4±0.4)×10
6 to (1.23±1.26)×10
6. Compared with the vehicle and ECC control groups, CAY10500 pretreatment did not obviously change the leucocytes ((11.24±6.13)×10
6,
P>0.05) and neutrophil counts ((2.27±2.20)×10
6,
P>0.05).
3.5. Pulmonary histopathology
As shown in Fig.
5a, although the alveoli retained their structures, there were significant alveolar congestion, hemorrhage, and infiltration of neutrophils in the airspace, and the thickness of the alveolar wall increased after ECC. Compared with the ECC control and vehicle groups, CAY10500 pretreatment did not change interstitial congestion, but significantly reduced the amount of neutrophils infiltrated (17.6±3.8/high-power field (HPF) vs. 22.4±3.3/HPF,
P<0.01; Fig.
5b).
Fig.5Lung tissue histology (a) and neutrophil counts (b) in experimental groups
Rats were treated as described in Fig.
2. Lung tissues were fixed in 10% formalin, dehydrated, embedded in paraffin, sectioned (3–5 μm), and then stained with hematoxylin-eosin (H&E). Specimens were examined under a light microscope. Neutrophils were calculated in 30 continuous areas 100 μm from the edge
3.6. Lung WW/DW ratio
The WW/DW ratio of lung was (84.2±1.7)% at baseline, and increased after ECC. There was no difference between the ECC control, vehicle, and CAY groups ((87.5±1.32)%, (88.2±1.11)%, and (87.9±1.49)%, respectively).
4. Discussion
Using an ALI model induced by rat ECC, we found that blocking the TNF-α binding to TNFR1 by CAY10500 inhibited neutrophil adhesion to the endothelial cells induced by TNF-α
in vitro, and pretreatment of CAY10500 intravenously
in vivo decreased the plasma TNF-α, but did not change the TNF-α levels in the lung tissue. As a result, CAY10500 slightly inhibited leukocyte recruitment and infiltration in the lung, but did not reduce lung edema or improve the lung function after ECC. Our results indicated that the TNFR1 pathway in circulating inflammatory cells played a limited role in ECC-induced ALI.
Other than small animal models as reported before (Wehberg et al.,
1996; Doguet et al.,
2004), the rat ECC model was applied to induce ALI in this study. The ECC with carotid artery-femoral vein cannulation did not need allogenic blood priming which could maintain Hb>70 g/L during ECC, and has the advantage of being an easy procedure to avoid disturbance to results caused by surgery (Du et al.,
2012). After 4 h of rest following 2 h of ECC procedure, TNF-α levels in plasma and BALF significantly increased, and alveolar congestion, hemorrhage, and infiltration of leukocytes in the airspace, and increased thickness of alveolar wall were seen in lung tissues, indicating that ALI was successfully induced by this ECC model.
The release of TNF-α initiated the release of IL-1, IL-8, and IL-6, as well as the up-regulation of TNF-α, which forms a positive inflammatory feedback (Allen et al.,
1992). Our results showed that TNF-α levels in both plasma and BAL are significantly increased after ECC, indicating TNF-α may mediate both systemic and pulmonary inflammation. By using CAY10500 pretreatment intravenously, TNF-α levels in the blood but not in the lung tissue were reduced. The result suggests that blocking of the circulating TNFR1 pathway inhibits the feedback of inflammation in the blood, but it cannot achieve a high enough concentration for inhibiting positive feedbacks of inflammatory cytokines in lung tissues.
TNF-α increases Mac-1 and intercellular adhesion molecule-1 (ICAM-1) expression, and subsequently induces neutrophil rolling on and adhesion to endothelial cells (Markovic et al.,
2009), followed by neutrophil accumulation and infiltration in the lung tissue. In our present study, with the increase of TNF-α following ECC, typical pulmonary inflammation was seen in the pulmonary pathology. Although our
in vitro study showed that blocking of the TNFR1 reduced TNF-α-induced neutrophil adhesion (Berg,
2006), intravenous pretreatment with CAY10500 inhibited leukocyte accumulation in the lung but did not attenuate ALI induced by ECC. The result suggests that TNFR1 expressed on circulating inflammatory cells mediates neutrophil-endothelial cell adhesion, but plays a limited role in mediating neutrophil migration and infiltration process.
During the migration and infiltration process, neutrophils need recognize chemoattractants to form leading edge and uropod tails (Vicente-Manzanares and Sanchez-Madrid,
2004; Burkhardt et al.,
2008; Du et al.,
2012). Therefore, the high chemokine concentration in the lung may play a more important role in localized lung inflammation (Muth et al.,
2005). In the present study, TNF-α levels in BALF were found significantly higher than those in blood (Khimenko et al.,
1999). The concentration gradient of TNF-α between blood and pulmonary interstitial tissue may mediate neutrophil migration and infiltration. However, TNF-α levels in pulmonary interstitial tissue cannot be blocked by CAY10500 intravenous treatment, which explains why the neutrophil infiltration was not inhibited by TNFR1 blocking.
Another reason for the negative results in this study is the complexity of ALI. Previous studies with CAY10500 were only performed with
in vitro cells, which avoided complicated cytokines and cell interactions. However, multiple cytokines form a very complicated inflammatory network
in vivo, which cannot be inhibited by only blocking TNFR1.
During ECC, local TNF-α in lung tissue may be an important chemoattractant supporting neutrophil infiltration. It was reported that intratracheal instillation of anti-TNF-α antibody attenuated cytomegalovirus (CMV) (Imai et al.,
1999) and ECC (Li et al.,
2013) induced lung injury. Therefore, intratracheal administration of CAY10500 might achieve a high enough concentration for inhibiting pulmonary inflammation.
In conclusion, although ECC can induce TNF-α release in lung tissue and blood, blocking TNFR1 by intravenous CAY10500 administration cannot inhibit TNF-α release or neutrophil accumulation in the lung. Our results indicated that the TNFR1 pathway in circulating inflammatory cell plays a limited role in ECC-induced ALI.
* Project supported by the National Natural Science Foundation of China (No. 81171651) and the Science and Technology Breakthrough Project of the Science and Technology Department of Sichuan Province (No. 2010FZ0044), ChinaCompliance with ethics guidelines Yu ZHAO, Chong-wei ZHANG, Wen-jing ZHOU, Jiao CHEN, Nan-fu LUO, Li-na GONG, Lei DU, and Jing ZHOU declare that they have no conflict of interest.References
[1] Allen, J.N., Herzyk, D.J., Allen, E.D., 1992. Human whole blood interleukin-1-β production: kinetics, cell source, and comparison with TNF-α.
J Lab Clin Med, 119(5):538-546.

[2] Berg, T., 2006. Inhibition of TNF-α signaling: divide and conquer.
ChemMedChem, 1(7):687-688.

[3] Burkhardt, J.K., Carrizosa, E., Shaffer, M.H., 2008. The actin cytoskeleton in T cell activation.
Annu Rev Immunol, 26(1):233-259.
[4] Carney, D.E., Lutz, C.J., Picone, A.L., 1999. Soluble tumor necrosis factor receptor prevents post-pump syndrome.
J Surg Res, 83(2):113-121.
[5] Doguet, F., Litzler, P.Y., Tamion, F., 2004. Changes in mesenteric vascular reactivity and inflammatory response after cardiopulmonary bypass in a rat model.
Ann Thorac Surg, 77(6):2130-2137.
[6] Drost, E.M., MacNee, W., 2002. Potential role of IL-8, platelet-activating factor and TNF-α in the sequestration of neutrophils in the lung: effects on neutrophil deformability, adhesion receptor expression, and chemotaxis.
Eur J Immunol, 32(2):393-403.
[7] Du, L., Zhou, J., Zhang, J., 2012. Actin filament re-organization is a key step in lung inflammation induced by systemic inflammatory response syndrome.
Am J Respir Cell Mol Biol, 47(5):597-603.
[8] He, M.M., Smith, A.S., Oslob, J.D., 2005. Small-molecule inhibition of TNF-α.
Science, 310(5750):1022-1025.
[9] Imai, Y., Kawano, T., Iwamoto, S., 1999. Intratracheal anti-tumor necrosis factor-α antibody attenuates ventilator-induced lung injury in rabbits.
J Appl Physiol, 87(2):510-515.
[10] Khimenko, P.L., Bagby, G.J., Fuseler, J., 1999. Tumor necrosis factor-α in ischemia and reperfusion injury in rat lungs.
J Appl Physiol, 85(6):2005-2011.
[11] Kotani, N., Hashimoto, H., Sessler, D.I., 2000. Neutrophil number and interleukin-8 and elastase concentrations in bronchoalveolar lavage fluid correlate with decreased arterial oxygenation after cardiopulmonary bypass.
Anesth Analg, 90(5):1046-1051.
[12] Li, T., Luo, N., Du, L., 2012. Early and marked up-regulation of TNF-α in acute respiratory distress syndrome after cardiopulmonary bypass.
Front Med, 6(3):296-301.
[13] Li, T., Luo, N., Du, L., 2013. Tumor necrosis factor-α plays an initiating role in extracorporeal circulation-induced acute lung injury.
Lung, 191(2):207-214.
[14] Markovic, N., McCaig, L.A., Stephen, J., 2009. Mediators released from LPS-challenged lungs induce inflammatory responses in liver vascular endothelial cells and neutrophilic leukocytes.
Am J Physiol Gastrointest Liver Physiol, 297(6):G1066-G1076.
[15] Mukhopadhyay, S., Hoidal, J.R., Mukherjee, T.K., 2006. Role of TNFα in pulmonary pathophysiology.
Respir Res, 7(1):125
[16] Muth, H., Kreis, I., Zimmermann, R., 2005. Differential gene expression in activated monocyte-derived macrophages following binding of factor VIIa to tissue factor.
Thromb Haemost, 94(5):1028-1034.
[17] Ng, C.S., Wan, S., Yim, A.P., 2002. Pulmonary dysfunction after cardiac surgery.
Chest, 121(4):1269-1277.
[18] Roach, D.R., Bean, A.G., Demangel, C., 2002. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection.
J Immunol, 168(9):4620-4627.
[19] Secher, T., Vasseur, V., Poisson, D.M., 2009. Crucial role of TNF receptors 1 and 2 in the control of polymicrobial sepsis.
J Immunol, 182(12):7855-7864.
[20] Sedgwick, J.D., Riminton, D.S., Cyster, J.G., 2000. Tumor necrosis factor: a master-regulator of leukocyte movement.
Immunol Today, 21(3):110-113.
[21] Staton, G.W., Williams, W.H., Mahoney, E.M., 2005. Pulmonary outcomes of off-pump vs on-pump coronary artery bypass surgery in a randomized trial.
Chest, 127(3):892-901.
[22] Tao, K., An, Q., Lin, K., 2009. Which is better to preserve pulmonary function: short-term or prolonged leukocyte depletion during cardiopulmonary bypass?.
J Thorac Cardiovasc Surg, 138(6):1385-1391.
[23] Vicente-Manzanares, M., Sanchez-Madrid, F., 2004. Role of the cytoskeleton during leukocyte responses.
Nat Rev Immunol, 4(2):110-122.
[24] Wehberg, K.E., Foster, A.H., Wise, R.M., 1996. Nitric oxide mediates fluid accumulation during cardiopulmonary bypass.
J Thorac Cardiovasc Surg, 112(1):168-174.
[25] Yende, S., Quasney, M.W., Tolley, E., 2003. Association of tumor necrosis factor gene polymorphisms and prolonged mechanical ventilation after coronary artery bypass surgery.
Crit Care Med, 31(1):133-140.
[26] Zhou, J., Luo, T., Rui, J., 2008. Changes of neutrophil functions after cardiopulmonary bypass: experiment with dogs.
Zhonghua Yi Xue Za Zhi, (in Chinese),88(17):1206-1210.
[27] Zhou, J., Wu, X.D., Lin, K., 2010. Blood hibernation: a novel strategy to inhibit systemic inflammation and coagulation induced by cardiopulmonary bypass.
Chin Med J (Engl), 123(13):1741-1747.



Open peer comments: Debate/Discuss/Question/Opinion
<1>