1. Introduction
MicroRNAs (miRNAs) are genome-encoded, about 21–23-nucleotide non-coding RNAs that negatively regulate gene expression by directing mRNA cleavage or interfering with translation (Carrington and Ambros,
2003). Studies have shown that miRNAs have the potential to regulate all physiological cell functions (Llave et al.,
2002). In plants, miRNAs regulate the expression of a large number of genes in diverse biological processes (Chen,
2004; Lu et al.,
2005; Zeng et al.,
2010; Shamimuzzaman and Vodkin,
2012; Zhang and Li,
2013), as identified by computational prediction, microarray analysis, and high-throughput sequencing (Jones-Rhoades and Bartel,
2004; Liu et al.,
2008; Zhao et al.,
2010; Zhu and Luo,
2013). Most miRNAs exist as independent transcription units, which are transcribed into long primary transcripts (pri-miRNAs) by RNA polymerase II, then cleaved to miRNA precursors (pre-miRNAs) (Bartel,
2004; Lee et al.,
2004). miRNAs have broad regulatory effects throughout the plant. Therefore, understanding the mechanisms regulating miRNA gene expression remains an important fundamental goal.
Class II promoters contain two parts, a core promoter and upstream elements. The core promoter consists of at least two elements: a TATA box, which begins at about the −30 bp position, and an initiator, which is centered on the transcription start site (TSS) (Liu et al.,
2010). Promoter elements of pre-miRNAs are similar to those of protein-coding genes (Smale,
2001). Bioinformatics analysis of the core miRNA promoters in model plants, including
Arabidopsis thaliana and
Oryza sativa, confirmed that the promoters of most known miRNA genes are of the same type as those of protein-coding genes and are transcribed by RNA polymerase II (Xie et al.,
2005; Zhou et al.,
2007; Cui et al.,
2009). TATA-less promoters were also identified in
Arabidopsis and rice. The promoters of most miRNA genes are found within 500-bp upstream of the TSS. Although substantial work has examined the promoters of protein-coding genes, the nature of miRNA promoter elements is still one of the most interesting aspects of small RNA biology (Megraw et al.,
2006).
Soybean (
Glycine max) is one of the most economically important agricultural crops in the world, and one of the most important global sources of protein and oil, both for food and for livestock feed. Soybean was the first legume species with a published high-quality draft genome sequence (Schmutz et al.,
2010). Because miRNAs play an important regulatory role in a wide variety of developmental and metabolic processes in plants, more and more soybean miRNAs have been identified (Zhang et al.,
2008; Chen et al.,
2009; Wang et al.,
2009; Guo et al.,
2011; Kulcheski et al.,
2011; Li et al.,
2011; Song et al.,
2011; Wong et al.,
2011; Zeng et al.,
2012; Hu et al.,
2013), but the features of the upstream sequences of these miRNAs have not been comprehensively analyzed. Core promoters and
cis-acting elements of 82 soybean miRNAs from the miRBase database were analyzed by bioinformatics methods (Liu et al.,
2010), but this analysis involved a relatively small number of miRNA genes. We have identified substantial numbers of miRNAs by deep sequencing of a
Glycine max degradome library (Hu et al.,
2013). In this study, the core promoters and
cis-acting elements of those miRNAs were predicted by bioinformatics methods to reveal the features of the core promoters of soybean miRNAs. The miRNA expression regulation pathway was also analyzed. These studies provide insights into the regulation of soybean miRNA expression.
2. Materials and methods
2.1. MicroRNA data and classification
A total of 440 miRNAs were identified from a soybean cDNA degradome library that was constructed from root, stem, leaf, and inflorescence tissues. The National Center for Biotechnology Information-Gene Expression Omnibus (NCBI-GEO) accession number of the
G.
max degradome sequences is GSE33380. All soybean miRNA sequences for prediction were divided into two groups based on their genomic background: “intronic miRNAs”, which overlap protein-coding genes, and “intergenic miRNAs”, which reside between protein-coding genes. The protein-coding genes included verified and hypothetical genes. The 440 soybean miRNAs included 34 intronic miRNAs and 406 intergenic miRNAs.
2.2. Identification of potential core promoters of soybean miRNAs
For promoter prediction, the upstream and downstream sequences of the 440 miRNA genes were downloaded from the soybean genome database (
http://www.phytozome.net/index.php) (Goodstein et al.,
2012). The upstream sequences of pre-microRNAs (hairpin precursors) of each miRNA gene were obtained according to Cui et al. (
2009). The 200-bp downstream sequence of each miRNA gene was also used for promoter prediction. For 83.86% of the pre-microRNAs (hairpin precursors), the upstream sequence was 2000 bp or longer. Promoters were predicted by the plant promoter identification program TSSP (
http://www.softberry.com), which is designed for predicting plant Pol II promoters (Shahmuradov et al.,
2005). The predictions were obtained at the default TSSP settings.
2.3. Analysis of cis-acting elements of soybean miRNAs
Potential promoter regions (from TSS to 800 bp upstream) were obtained to predict the potential
cis-acting elements and motifs. If there were overlapping TSSs in the selected region, only the sequence between the two TSSs was analyzed to exclude redundancy (Liu et al.,
2010). The PlantCARE database (
http://bioinformatics.psb.ugent.be/webtools/plantcare/html), a database of plant
cis-acting regulatory elements (Lescot et al.,
2002), was used to analyze the
cis-acting elements of the miRNAs.
3. Results
3.1. Core promoters and enhancers of soybean miRNAs
Putative promoters were divided into TATA-box/TSS core promoters and enhancers, and counted independently. From 440 miRNAs, we identified 699 predicted core promoters, with 102 predicted enhancers in upstream sequences. There were 38 core promoters, but no enhancers, predicted in the downstream sequence.
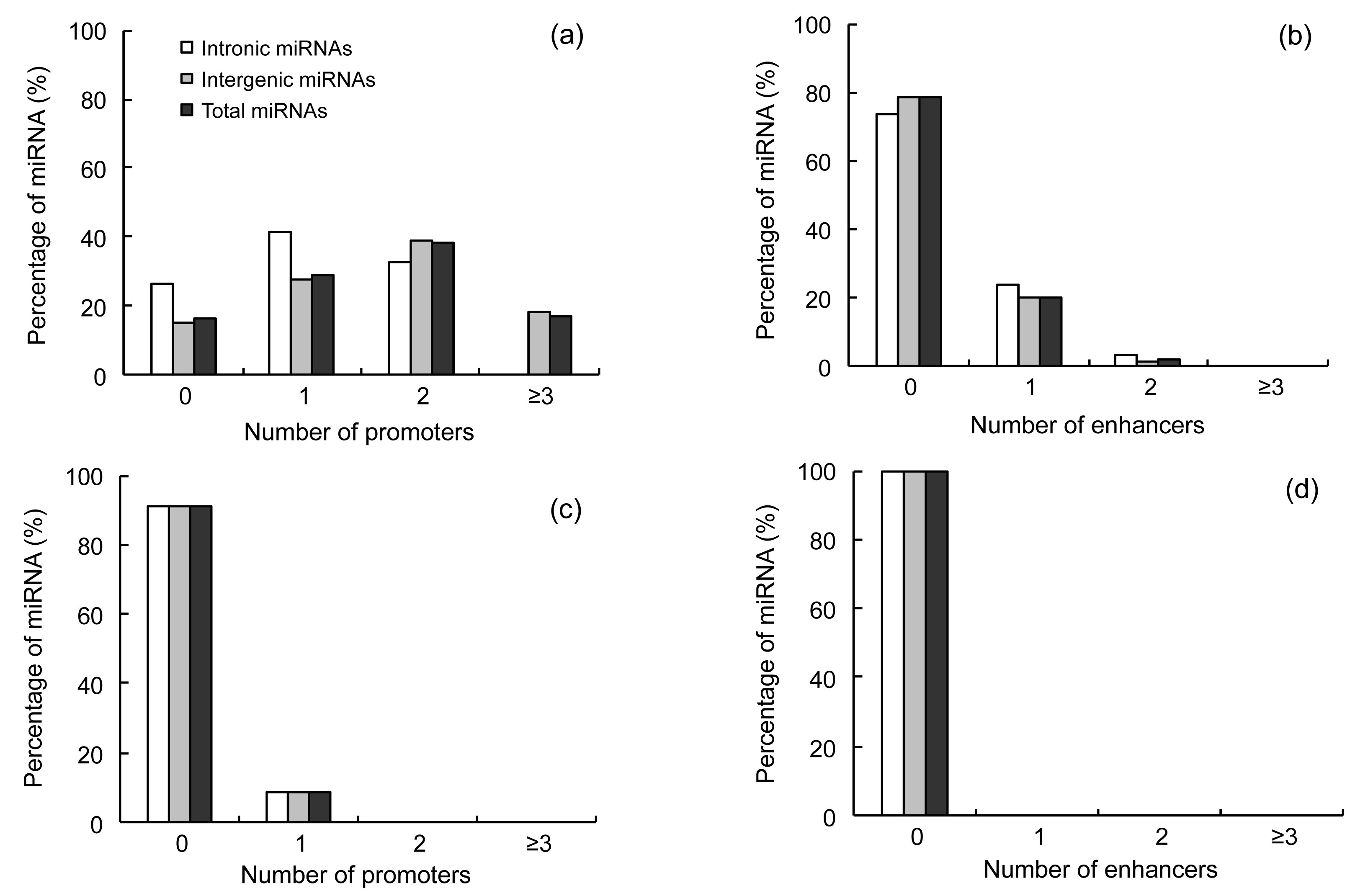
Among the upstream sequences of the 440 miRNAs, 71 (16.14%) were found to contain no predicted promoters (Fig.
1a). Of the other 369 (83.86%) promoter-containing sequences, 127 (28.86%) were predicted to have only one promoter, 168 (38.18%) contained two, and the rest, 74 (16.90%), contained three or more predicted promoters. Among the predicted enhancers in upstream sequences of the 440 miRNAs, 345 (78.41%) were found to contain no enhancers (Fig.
1b). Among the other 95 (21.59%) enhancer-containing sequences, 88 (20.00%) were predicted to have only one enhancer, 7 (1.59%) contained two enhancers, and no sequences were predicted to contain three or more enhancers. Most pre-miRNAs examined contained no predicted enhancers.
Fig.1
Distribution of numbers of promoters in upstream (a) and downstream (c) sequences of miRNAs and distribution of numbers of enhancers in upstream (b) and downstream (d) sequences of miRNAs
In the downstream sequences of the 440 miRNAs, 402 (91.36%) were found to contain no predicted promoter (Fig.
1c) and 38 (8.64%) were predicted to have only one. None of the 440 miRNAs contained predicted enhancers in the downstream sequence (Fig.
1d).
Pre-miRNAs from intergenic regions generally had more core promoters than the intronic pre-miRNAs (Table
S1), and two or fewer core promoters were identified for each intronic miRNA. In the pre-miRNA downstream sequences, there were few core promoters, and never more than one.
3.2. Distribution of the predicted core promoters and enhancers of soybean miRNA genes
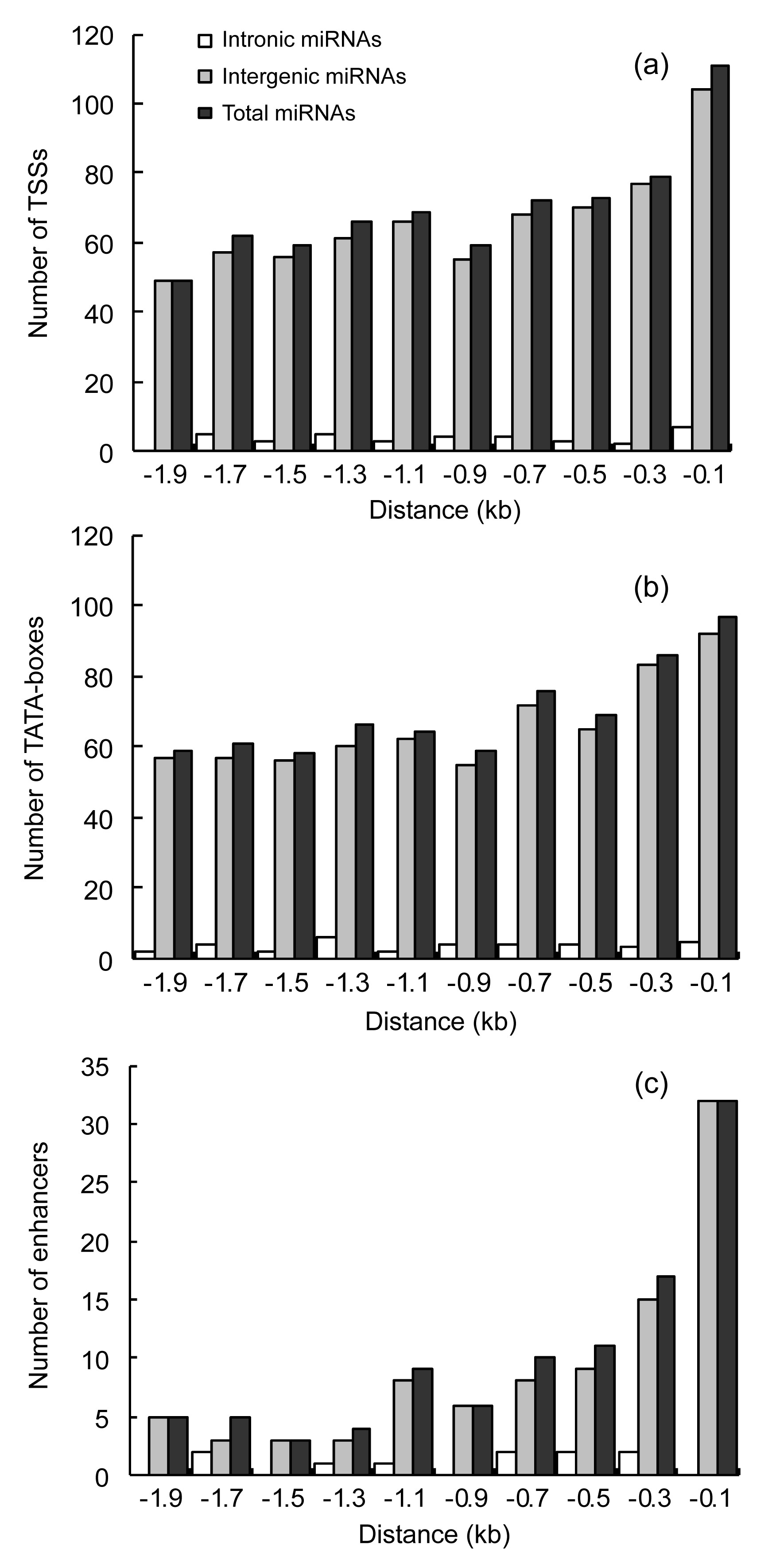
We analyzed the specific sequences of pre-miRNAs using the TSSP program, and counted the TSS, TATA-box, and enhancers separately. For the predicted core promoters in the upstream sequences of the 440 miRNAs, a total of 699 TSSs were identified. A strong peak was observed near the start position of each transcript. For intergenic miRNAs, the distribution could be separated into three distinct regions (Fig. 2a). The first peak was found close to the pre-miRNA within the 1.0-kb upstream region, which contained 56.41% of the total predicted TSSs (663). A second broad region, from −1.0 to −1.6 kb, contained 27.60% of the predicted TSSs. The third region, from −1.6 to −2.0 kb, contained 15.99% of the predicted TSSs. For intronic miRNAs, there were also three peaks of TSSs (from 0 to −1.2 kb, −1.2 to −1.6 kb, and −1.6 to −2.0 kb).
Fig.2
Genomic distributions of TSSs (a), TATA-boxes (b), and enhancers (c) from the 5′ end of the pre-miRNA upstream sequence
The distribution of TATA-boxes was similar to the distribution of TSSs. The core promoters of four miRNAs (gm-cmiR160f, gm-cmiR319k, gm-cmiR13, and gm-cmiR4i) were found to be TATA-less. For intergenic miRNAs, we identified three distinct regions (Fig. 2b). The first peak was found close to the pre-miRNA within the 0.6-kb upstream region, and contained 36.42% of the total (659) predicted TATA-boxes. A second broad region, from −0.6 to −1.2 kb, contained 28.68% of predicted TATA-boxes. The third region, from −1.1 to −2.0 kb, contained 34.90%. For intronic miRNAs, there were also three peaks of TATA-boxes (from 0 to −0.6 kb, −0.6 to −1.2 kb, and −1.2 to −2.0 kb).
In intergenic and intronic miRNAs, 102 enhancers were predicted. For intergenic miRNAs, most (74.51%) of the predicted enhancers were found within 1.0-kb upstream of pre-miRNAs. The remaining 15.69% occurred within −1.0 to −1.6 kb. The third region, from −1.6 to −2.0 kb, contained 9.80% of the predicted enhancers (Fig. 2c). For the predicted core promoters in downstream sequences of the 440 miRNAs, a total of 36 (8.18%) TSSs and TATA-boxes were identified.
3.3.
Cis-acting elements in potential promoters and an miRNA expression regulation pathway related to hormone metabolism
To further elucidate the spatiotemporal expression pattern and the function of miRNAs from the G. max degradome library, 800 bp upstream sequences of the potential TSS of 369 miRNA genes were analyzed using the PlantCARE database, excluding 71 miRNAs that had no predicted TSS. Cis-acting elements and motifs of 355 miRNA promoters were predicted, excluding 14 miRNAs without sufficient sequence information. There were 126 types of cis-acting elements. Every miRNA contained a TATA-box core promoter element at around −30 bp from the TSS and a CAAT-box, a common cis-acting element, in the promoter and enhancer regions (Table S2). In addition, 99.72% of the miRNAs contained a light responsive element (LRE). Cis-acting elements were grouped into several classes according to their functions, such as stress-responsive elements (SREs), cis-acting elements regulating plant growth (GREs), hormone-regulated cis-acting elements (HREs), and tissue-specific cis-acting elements (TSEs) (Table S3) as described by Liu et al. (2010). Other regulatory elements, such as AT-rich DNA, cis-acting regulatory elements essential for anaerobic induction, and elicitor-responsive elements, were not classified. Of the miRNA sequences examined, 86.48% contained an SRE, including anaerobic induction elements (AREs), defense SREs (TC-rich repeats), ABA (abscisic acid)-response elements (ABREs), low-temperature responsive elements (LTRs), MYB (v-myb avian myeloblastosis viral oncogene homolog) binding sites involved in drought response (MBSs), salicylic acid responsiveness elements (SAEs), heat-stress responsive elements (HSEs), and LREs. Also, 35.49% of miRNAs contained a GRE, including a leaf morphology development element (HD-Zip 2), differentiation of the palisade mesophyll cells element (HD-Zip 1), meristem-specific activation elements (OCT, NON-box, dOCT, CCGTCC-box, CAT-box), zein metabolism regulation element (O2-site), circadian control element (circadian), and cell cycle regulation elements (MSA-like). TSEs were identified in 72.39% of miRNA sequences examined, including nodule-specific factor binding site (Nodule-site2), involved in the endosperm-specific negative expression element (AACA_motif), endosperm expression elements (GCN4_motif, Skn-1_motif), and cis-acting regulatory elements involved in root-specific expression (as1) and seed-specific regulation (RY-element). HREs were found in 70.70% of miRNA sequences examined, including those possibly involved in the regulation of the response to plant hormones, such as gibberellin (GA), auxin-responsive element, ethylene, salicylic acid, methyl jasmonate, and flavonoids.
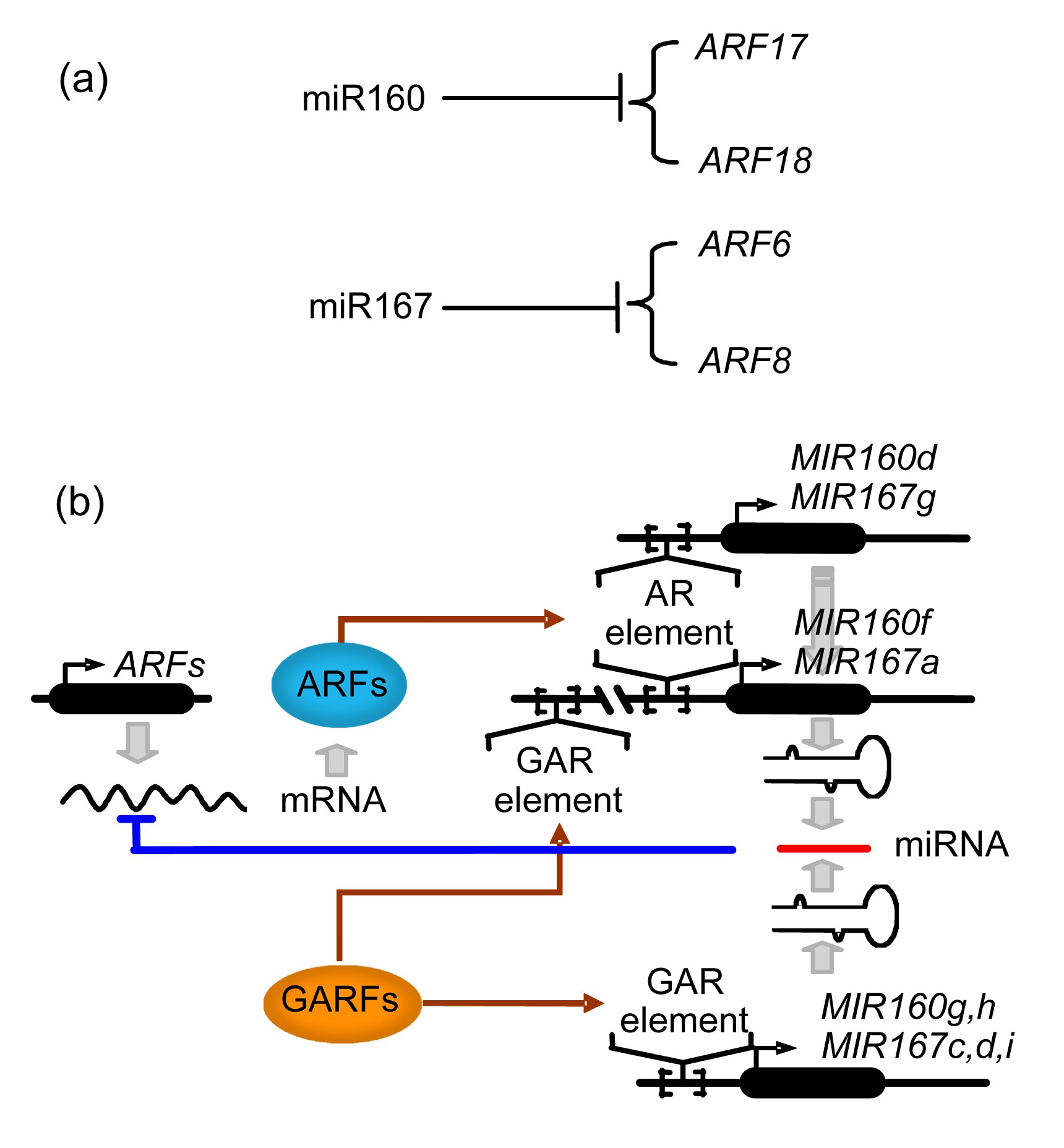
Among HREs, numerous copies of the auxin response factor (ARF) elements are found in the putative promoter regions of MIR160d, MIR160f, MIR167a, and MIR167g. Numerous copies of gibberellin response factor (GARF) elements are more frequently found in the putative promoter regions of MIR160f, MIR160g, MIR160h, MIR167a, MIR167c, MIR167d, and MIR167i. In our miRNA target gene data, multiple ARF family members, including ARF17, ARF18, ARF6, and ARF8, were predicted to be directly targeted by miR160 and miR167. Based on the above data, we propose a possible ARF- and GARF-mediated negative feedback regulatory loop involving miR160, miR167, and certain ARF family members (Fig. 3). ARF17 and ARF18 are post-transcriptionally regulated by miR160, and ARF6 and ARF8 are regulated by miR167 (Fig. 3a). In the negative feedback regulatory loop, miR160 and miR167 repress their target ARF, and ARFs affect miR160 and miR167 accumulation via binding to the AR regulation elements of the MIR160 and MIR167 promoters. In addition to the feedback regulation of miR160 and miR167 by ARF, GARF can regulate miR160 and miR167 via binding to the GARF elements of the MIR160 and MIR167 promoters (Fig. 3b). According to the feedback regulation of miR160 and miR167 by ARF and GARF, we speculate that there is GA-auxin cross-talk in the pathway of the regulation of miRNA expression.
Fig.3
An speculated miRNA expression regulation pathway related to hormone metabolism
(a) Certain auxin response factor (ARF) family members are directly regulated by miRNAs (miR160 and miR167). (b) A possible negative feedback regulatory loop involving miR160, miR167, and certain ARF family members. Numerous copies of the ARF recognition motif are found in the putative promoter regions of MIR160d, MIR160f, MIR167a, and MIR167g. Also, numerous copies of gibberellin (GA) response (GAR) elements are found in the putative promoter regions of MIR160f, MIR160g, MIR160h, MIR167a, MIR167c, MIR167d, and MIR167i. We speculate that GA regulates miRNAs to inhibit the formation of auxin
4. Discussion
The core promoters and cis-acting elements of 440 miRNAs from a G. max degradome library were analyzed using bioinformatics methods to reveal the general features of the promoters of a large number of soybean miRNAs, examine their cis-acting elements, and provide insights into the transcriptional regulation and functions of miRNAs in soybean. Recent studies of Arabidopsis and rice showed that most core promoters contain a TATA-box (Megraw et al., 2006; Cui et al., 2009), and our results for soybean support this observation. Among 699 core promoters predicted, only 4 (0.57%) were TATA-less promoters. Also, 95 of 440 miRNAs (21.59%) contained enhancers. The promoters predicted by bioinformatics analysis will offer valuable information for the design of verification assays and for functional research on miRNAs.
The expression of genes at the transcriptional and post-transcriptional levels is often controlled by small cis-acting sequence elements in or near the regulated genes. Cis-acting elements have been analyzed extensively (Zhou et al., 2007; Liu H.H. et al., 2008; Liu Y.X. et al., 2010). In this study, cis-acting elements of 369 soybean miRNAs were predicted and were classified into several classes according to their functions, such as SRE, GRE, HRE, and TSE. Among those cis-acting elements, HRE sites were further analyzed to reveal the potential functional correlation between the transcription factors, miRNAs, and the target genes of the miRNAs. In Arabidopsis, the ARF element was found to be over-represented among binding motifs in miRNA promoters. MicroRNAs with putative ARF binding sites upstream may play a role in negative feedback loops that control their own expression (Jones-Rhoades et al., 2006; Megraw et al., 2006; Xie et al., 2010). In soybean, we also identified ARF elements in some miRNA promoters, such as miR160 and miR167. Multiple ARF family members, including ARF17, ARF18, ARF6, and ARF8, were predicted to be directly targeted by miR160 and miR167. Although the target genes of soybean miRNAs have not been experimentally verified, targeting of ARFs by soybean miRNAs was indicated by experimental results showing that Arabidopsis miR160 and miR167 target ARFs (Mallory et al., 2005; Wang et al., 2005; Wu et al., 2006; Liu et al., 2007). According to the negative feedback model, we propose a possible ARF-mediated negative feedback regulatory loop in soybean, which indicates that miRNAs often act as regulators with many secondary effects on gene expression. We also identified many types of GARF-binding motifs in promoters of MIR160 and MIR167. Gou et al. (2010) found that gibberellins regulate lateral root formation in Populus through interacting with auxin, and auxin levels increased in GA-deficient and insensitive transgenic roots, suggesting crosstalk between GA signaling and other hormone pathways. GA may negatively regulate auxin, but the genes and pathways responsible have remained unclear. Here, we found GARF-binding motifs in the promoters of MIR160 and MIR167 and propose a GARF-mediated negative feedback regulatory loop for soybean miRNA expression, suggesting that gibberellins regulate the expression of miR160 and miR167 to interplay with auxin. The functions of these computationally identified cis-acting elements of soybean miRNA will require experimental validation.
Although few soybean miRNAs have been studied thoroughly, the multiple types of cis-acting elements we have identified and the predicted target genes of these soybean miRNAs both indicate that soybean miRNAs have diverse regulatory functions. For example, the gma-miR156 family has 71 types of cis-acting elements and regulates 27 target genes. Moreover, the more members in each miRNA family, the more genes the miRNAs target, and the more types of cis-acting elements are contained in the promoter of each miRNA.
In summary, we have performed a bioinformatics analysis of the core promoter sequences (TSS, TATA-box, and enhancer) of soybean pre-miRNAs (upstream and downstream sequences) from a G. max degradome library. A total of 699 promoters and 102 enhancers for 440 soybean pre-miRNAs were detected. Most miRNA genes were intergenic and there were more core promoters in intergenic genes than in intronic genes. Distribution analysis showed that the locations of the TATA-box and TSS were similar. There were 126 types of cis-acting elements identified in 355 miRNA promoters, including SREs, GREs, HREs, and TSEs. In addition, we suggest an ARF- and GARF-mediated negative feedback regulatory loop for soybean miRNA expression. These findings improve our understanding of the specific sequences and motifs upstream of pre-miRNAs in soybean. They also provide a basis for further research on the functional role of miRNA and the regulation of miRNA expression in soybean.
* Project supported by the National Natural Science Foundation of China (No. 31271652) and the National Science Foundation for Young Scientists of China (No. 31301708)# Electronic supplementary materials: The online version of this article (http://dx.doi.org/10.1631/jzus.B1300179) contains supplementary materials, which are available to authorized usersCompliance with ethics guidelines Yi-qiang HAN, Zheng HU, Dian-feng ZHENG, and Ya-mei GAO declare that they have no conflict of interest.
References
[1] Bartel, D.P., 2004. MicroRNAs: genomics, biogenesis, mechanism, and function.
Cell, 116(2):281-297.


[2] Carrington, J.C., Ambros, V., 2003. Role of microRNAs in plant and animal development.
Science, 301(5631):336-338.
[3] Chen, R., Hu, Z., Zhang, H., 2009. Identification of microRNA in wild soybean (
Glycine soja).
J Integr Plant Biol, 51(12):1071-1079.
[4] Chen, X., 2004. A microRNA as a translational repressor of APETALA2 in
Arabidopsis flower development.
Science, 303(5666):2022-2025.
[5] Cui, X., Xu, S.M., Mu, D.S., 2009. Genomic analysis of rice microRNA promoters and clusters.
Gene, 431(1-2):61-66.
[6] Goodstein, D.M., Shu, S., Howson, R., 2012. Phytozome: a comparative platform for green plant genomics.
Nucl Acids Res, 40(D1):D1178-D1186.
[7] Gou, J., Strauss, S.H., Tsai, C.J., 2010. Gibberellins regulate lateral root formation in
Populus through interactions with auxin and other hormones.
Plant Cell, 22(3):623-639.
[8] Guo, N., Ye, W.W., Wu, X.L., 2011. Microarray profiling reveals microRNAs involving soybean resistance to
Phytophthora sojae
.
Genome, 54(11):954-958.
[9] Hu, Z., Jiang, Q., Ni, Z., 2013. Analyses of a
Glycine max degradome library identify microRNA targets and microRNAs that trigger secondary siRNA biogenesis.
J Integr Plant Biol, 55(2):160-176.
[10] Jones-Rhoades, M.W., Bartel, D.P., 2004. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA.
Mol Cell, 14(6):787-799.
[11] Jones-Rhoades, M.W., Bartel, D.P., Bartel, B., 2006. MicroRNAs and their regulatory roles in plants.
Ann Rev Plant Biol, 57(1):19-53.
[12] Kulcheski, F.R., de Oliveira, L.F., Molina, L.G., 2011. Identification of novel soybean microRNAs involved in abiotic and biotic stresses.
BMC Genomics, 12(1):307
[13] Lee, Y., Kim, M., Han, J., 2004. MicroRNA genes are transcribed by RNA polymerase II.
EMBO J, 23(20):4051-4060.
[14] Lescot, M., Dehais, P., Thijs, G., 2002. PlantCARE, a database of plant
cis-acting regulatory elements and a portal to tools for
in silico analysis of promoter sequences.
Nucl Acids Res, 30(1):325-327.
[15] Li, H., Dong, Y., Yin, H., 2011. Characterization of the stress associated microRNAs in
Glycine max by deep sequencing.
BMC Plant Biol, 11(1):170
[16] Liu, H.H., Tain, X., Li, Y.J., 2008. Microarray-based analysis of stress-regulated microRNAs in
Arabidopsis thaliana
.
RNA, 14(5):836-843.
[17] Liu, P.P., Montgomery, T.A., Fahlgren, N., 2007. Repression of
AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages.
Plant J, 52(1):133-146.
[18] Liu, Y.X., Han, Y.P., Chang, W., 2010. Genomic analysis of microRNA promoters and their
cis-acting elements in soybean.
Agric Sci China, 9(11):1561-1570.
[19] Llave, C., Xie, Z., Kasschau, K.D., 2002. Cleavage of
Scarecrow-like mRNA targets directed by a class of
Arabidopsis miRNA.
Science, 297(5589):2053-2056.
[20] Lu, S., Sun, Y.H., Shi, R., 2005. Novel and mechanical stress-responsive microRNAs in
Populus trichocarpa that are absent from
Arabidopsis
.
Plant Cell, 17(8):2186-2203.
[21] Mallory, A.C., Bartel, D.P., Bartel, B., 2005. MicroRNA-directed regulation of
Arabidopsis AUXIN RESPONSE FACTOR17 is essential for proper development and modulates expression of early auxin response genes.
Plant Cell, 17(5):1360-1375.
[22] Megraw, M., Baev, V., Rusinov, V., 2006. MicroRNA promoter element discovery in
Arabidopsis
.
RNA, 12(9):1612-1619.
[23] Schmutz, J., Cannon, S.B., Schlueter, J., 2010. Genome sequence of the palaeopolyploid soybean.
Nature, 463(7278):178-183.
[24] Shahmuradov, I.A., Solovye, V.V., Gammerman, A.J., 2005. Plant promoter prediction with confidence estimation.
Nucl Acids Res, 33(3):1069-1076.
[25] Shamimuzzaman, M., Vodkin, L., 2012. Identification of soybean seed developmental stage-specific and tissue-specific miRNA targets by degradome sequencing.
BMC Genomics, 13(1):310
[26] Smale, S.T., 2001. Core promoters: active contributors to combinatorial gene regulation.
Genes Dev, 15(19):2503-2508.
[27] Song, Q.X., Liu, Y.F., Hu, X.Y., 2011. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing.
BMC Plant Biol, 11(1):5
[28] Wang, J.W., Wang, L.J., Mao, Y.B., 2005. Control of root cap formation by microRNA-targeted auxin response factors in
Arabidopsis
.
Plant Cell, 17(8):2204-2216.
[29] Wang, Y., Li, P., Cao, X., 2009. Identification and expression analysis of miRNAs from nitrogen-fixing soybean nodules.
Biochem Biophys Res Commun, 378(4):799-803.
[30] Wong, C.E., Zhao, Y.T., Wang, X.J., 2011. MicroRNAs in the shoot apical meristem of soybean.
J Exp Bot, 62(8):2495-2506.
[31] Wu, M.F., Tian, Q., Reed, J.W., 2006.
Arabidopsis microRNA167 controls patterns of
ARF6 and
ARF8 expression, and regulates both female and male reproduction.
Development, 133(21):4211-4218.
[32] Xie, Z., Allen, E., Fahlgren, N., 2005. Expression of
Arabidopsis MIRNA genes.
Plant Physiol, 138(4):2145-2154.
[33] Xie, Z.X., Khanna, K., Ruan, S., 2010. Expression of microRNAs and its regulation in plants.
Semin Cell Dev Biol, 21(8):790-797.
[34] Zeng, H.Q., Zhu, Y.Y., Huang, S.Q., 2010. Analysis of phosphorus-deficient responsive miRNAs and
cis-elements from soybean (
Glycine max L.).
J Plant Physiol, 167(15):1289-1297.
[35] Zeng, Q.Y., Yang, C.Y., Ma, Q.B., 2012. Identification of wild soybean miRNAs and their target genes responsive to aluminum stress.
BMC Plant Biol, 12(1):182
[36] Zhang, B., Pan, X., Stellwag, E.J., 2008. Identification of soybean microRNAs and their targets.
Planta, 229(1):161-182.
[37] Zhang, Y., Li, Y.K., 2013. MicroRNAs in the regulation of immune response against infections.
J Zhejiang Univ-Sci B (Biomed & Biotechnol), 14(1):1-7.
[38] Zhao, C.Z., Xia, H., Frazier, T.P., 2010. Deep sequencing identifies novel and conserved microRNAs in peanuts (
Arachis hypogaea L.).
BMC Plant Biol, 10(1):3
[39] Zhou, X., Ruan, J., Wang, G., 2007. Characterization and identification of microRNA core promoters in four model species.
PLoS Comput Biol, 3(3):e37
[40] Zhu, Q.W., Luo, Y.P., 2013. Identification of miRNAs and their targets in tea (
Camellia sinensis).
J Zhejiang Univ-Sci B (Biomed & Biotechnol), 14(10):916-923.
Open peer comments: Debate/Discuss/Question/Opinion
<1>