1. Introduction
Geminiviruses are a group of circular, single-stranded DNA viruses that cause devastating diseases worldwide. Four genera (
Mastrevirus,
Begomovirus,
Curtovirus, and
Topocuvirus) have been taxonomically classified within the family Geminiviridae, with the genus
Begomovirus being the largest, containing more than 200 species. Earlier studies have demonstrated that mutation, recombination, and pseudo-recombination are the major contributory factors driving the emergence of begomovirus variants (Czosnek and Ghanim,
2011). Although geminiviruses depend on host DNA polymerase for replication, mutation occurs frequently in geminivirus genomes and progeny populations of high variability can be generated in a short period of time. Mean rates of 1.60×10
−3 and 1.33×10
−4 nucleotide substitutions per site per year (subs/site/year) have been estimated for the DNA-A and DNA-B components, respectively, of the East African cassava mosaic virus (EACMV) (Duffy and Holmes,
2009). Similar rates of substitution have also been reported for artificially inoculated hosts (Ge et al.,
2007). It is now clear that geminiviruses evolve as rapidly as many RNA viruses, but only a few reports estimate the time required to generate such levels of genetic variability.
Tomato yellow leaf curl virus (TYLCV) is a monopartite begomovirus belonging to the genus
Begomovirus, and is transmitted by the whitefly
Bemisia tabaci in a circulative and persistent manner. The genome of TYLCV encodes two partially overlapping genes in the viral strand:
V1 encodes a coat protein, and
V2 encodes a precoat protein and acts as a suppressor of RNA silencing (Glick et al.,
2008; Jiang et al.,
2012). The complementary strand of TYLCV encodes four proteins: a replication-associated protein (C1/Rep), a transcriptional activator protein (C2/TrAP), a replication enhancer protein (C3/REn), and protein C4, which is entirely embedded in the
C1 gene, but has a different reading frame. Open reading frames (ORFs) are organized bi-directionally and are separated by an intergenic region (IR) which serves as the origin of viral replication and as a bi-directional promoter (Hanley-Bowdoin et al.,
1999).
Tomato yellow leaf curl disease (TYLCD) caused by TYLCV is becoming the most destructive disease in tomato plants (Moriones and Navas-Castillo,
2000). Plants infected by TYLCV during the early growth stages frequently suffer yield losses of up to 100%. Although tomato plants in Middle East countries have been severely affected by TYLCV only since the 1960s, the first report of damage caused by this disease dates back to 1931 in Israel (Cohen and Antignus,
1994). Since then, epidemics have emerged and devastated tomato crops rapidly and widely. TYLCV was first reported in Shanghai, China in 2006 (Wu et al.,
2006), and it has since spread rapidly to 13 provinces or autonomous regions of China, including Guangdong, Zhejiang, Jiangsu, Anhui, Shandong, Hebei, Henan, Beijing, Tianjin, Shanxi, Shaanxi, and Inner Mongolia and Xinjiang Uygur Autonomous Regions (Zhang et al.,
2009). Since no other begomoviruses had been detected in Shanghai before 2006, the rapid spread of TYLCV provided a great opportunity to understand the evolution of a plant DNA virus soon after its first introduction to a new area. Twenty-six TYLCV isolates were collected from the same area of Shanghai during 2006–2010. Full-length genomic sequencing allows for a more comprehensive analysis of virus variability and of the different forces driving the evolution of TYLCV.
2. Materials and methods
2.1. Collection of TYLCV-infected tomato plants
Twenty-six tomato plants (cultivar Pufen 1, formerly called 98-8) with typical TYLCV symptoms (leaf curling, yellowing, and stunted growth) were collected from Shanghai from 2006 to 2010. TYLCV isolates were named after the year in which they were collected (Table
1).
Table 1
TYLCV isolates collected in Shanghai, China during 2006–2010
| Isolate |
Collection date |
| SH2006-1 |
2006 |
| SH2006-2 |
2006 |
| SH2007-1 |
2007 |
| SH2007-2 |
2007 |
| SH2007-3 |
2007 |
| SH2007-4 |
2007 |
| SH2007-5 |
2007 |
| SH2007-6 |
2007 |
| SH2008-1 |
2008 |
| SH2008-2 |
2008 |
| SH2008-3 |
2008 |
| SH2008-4 |
2008 |
| SH2008-5 |
2008 |
| SH2008-6 |
2008 |
| SH2009-1 |
2009 |
| SH2009-2 |
2009 |
| SH2009-3 |
2009 |
| SH2009-4 |
2009 |
| SH2009-5 |
2009 |
| SH2009-6 |
2009 |
| SH2010-1 |
2010 |
| SH2010-2 |
2010 |
| SH2010-3 |
2010 |
| SH2010-4 |
2010 |
| SH2010-5 |
2010 |
| SH2010-6 |
2010 |
2.2. Genomic DNA extraction and detection of TYLCV
Total genomic DNA was extracted from 0.5 g of leaf tissue using the cetyltrimethylammonium bromide (CTAB) method (Zhou et al.,
2001). After washing in 70% ethanol, the DNA pellets were dried at room temperature and re-suspended in about 200 μl of distilled water. Primers TYLCV/F (5'-GAGCTCTTAGCTGCCTGAATGTTC-3') and TYLCV/R (5'-GAGCTCAACAGATGTCAAGACCTAC-3'), which were expected to specifically amplify a 2.8-kb fragment of TYLCV, were used to detect TYLCV infection.
2.3. Isolation, cloning, and sequencing of full-length genomes
Rolling circle amplification (RCA) based on the phi29 DNA polymerase available in the TempliPhi™ kit (GE Healthcare, UK) was used to amplify circular viral genomes from 1 μg of total plant DNA extractions (Guo et al.,
2009). The amplified DNA concatemers (5 μl) were cleaved with
SacI to generate full-length viral fragments, which were subsequently inserted into linearized pGEM-7Zf+ (Promega, Madison, WI). About five independent clones from each sample were randomly chosen for sequencing by an automated model 3730 DNA sequencer (Perkin-Elmer, USA) using M13 forward and reverse primers and a pair of walking primers, TYLCV-W/F (5'-TCTGCAATCCAGGACCTACC-3') and TYLCV-W/R (5'-AGTCTATCTTGCAATATGTG-3'). Sequences were assembled and edited using EditSeq (Lasergene Package of Programs from DNASTAR Inc.). The fidelity of phi29 DNA polymerase was pre-determined by sequencing 20 individual clones generated directly from the TYLCV infectious clone using this enzyme during the initial amplification trials, and the mutation frequency was estimated to be 1.8×10
−5.
2.4. Sequence analysis
The circular genomic sequences were arranged to begin at the nick-site in the invariant nonanucleotide sequence at the origin of replication (TAATATT-3'/5'-AC) (Padidam et al.,
1995). Multiple sequence alignment was generated with the aid of ClustalX 1.83 (Thompson et al.,
2002). The level of genetic variation was calculated by estimating Watterson’s estimator of
θ and the average number of nucleotide differences per site between two sequences (Pi) using DnaSP v5 (Guo et al.,
2008; Librado and Rozas,
2009). The ratio of pairwise nonsynonymous (
d
n) to synonymous (
d
s) nucleotide substitutions per site, was estimated using the Pamilo-Bianchi-Li method implemented in MEGA 5 (Tamura et al.,
2011). Confidence estimates for nonsynonymous and synonymous nucleotide substitutions were assessed by the bootstrap method based on 100 replicates. The extent of mutational bias and the number of unambiguous nucleotide changes of each type were calculated by MEGA 5 (Tamura et al.,
2011). Genetic distances within and between subpopulations were calculated by applying Kimura’s two-parameter method in MEGA 5 (Tamura et al.,
2011). Evolutionary constraints acting on the overlapping regions were assessed using entropy values and BioEdit software (Hall,
1999). Cumulative synonymous and nonsynonymous mutations were determined using Synonymous Non-synonymous Analysis Program (SNAP) (
http://www.hiv.lanl.gov/content/sequence/SNAP/SNAP.html) as described by Korber (
2002) and Melgarejo et al. (
2013).
3. Results
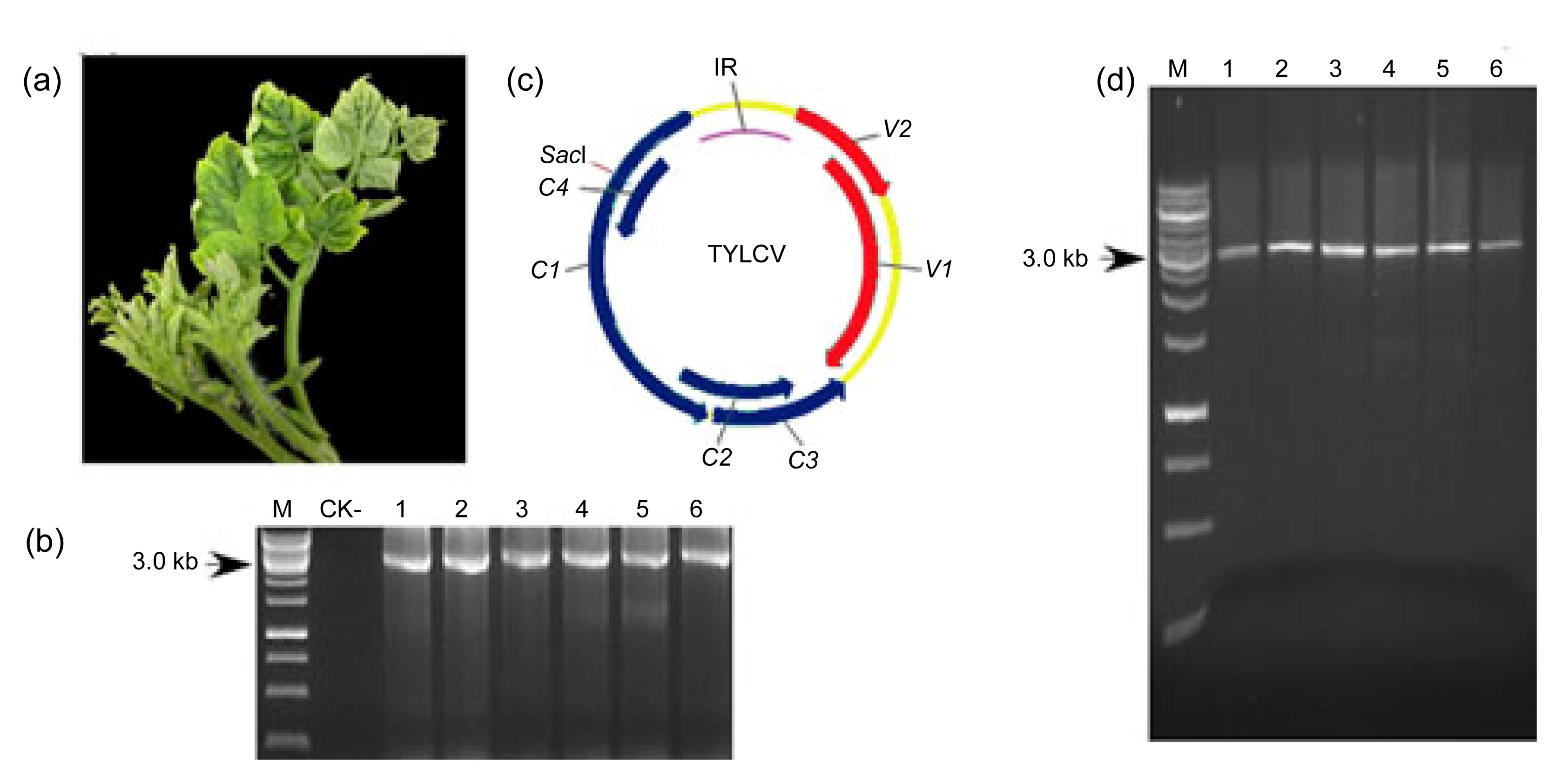
3.1. Symptom observation and detection of TYLCV
From 2006 to 2010, a total of 26 samples were obtained from surveys conducted once a year in Shanghai, China. All the plants sampled showed typical TYLCV symptoms, such as leaf curling, yellowing, and stunted growth (Fig.
1a). The specific 2.8-kb DNA fragment was amplified by polymerase chain reaction (PCR) from all the samples using primers TYLCV/F and TYLCV/R (Fig.
1b), indicating that these symptomatic tomato plants were infected by TYLCV. To obtain the complete nucleotide sequence of each of the 26 isolates, RCA followed by
SacI digestion of the DNA concatemers was performed to obtain the full-length viral fragment (Fig.
1d). About five independent clones from each sample were chosen to be sequenced and were found to be 2781 nucleotides (nts) long with a genomic organization typical of begomoviruses (Fig.
1c).
Fig.1
Genome composition and detection of TYLCV-infected virus samples
(a) TYLCV-infected tomato plants showing leaf curling, yellowing, and stunted growth symptoms. (b) PCR detection of TYLCV in six of the infected tomato plants (target size, 2.8 kb). (c) Genomic organization of TYLCV. The intergenic region and six ORFs encoded by the viral strand and complementary strand are shown as indicated. (d) Full-length TYLCV fragment obtained from SacI digestion of rolling circle amplification products. IR: intergenic region; M: DNA marker
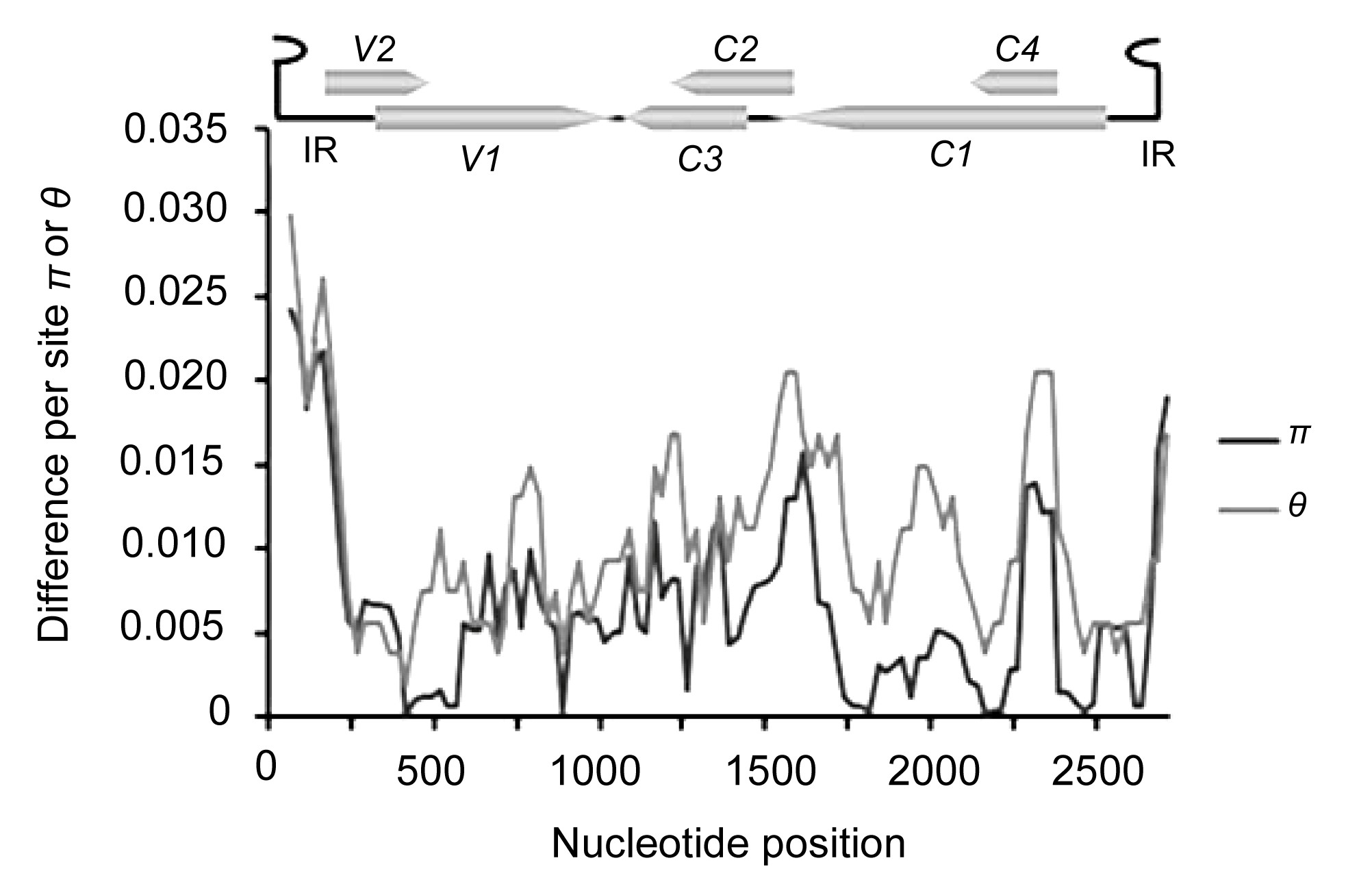
3.2. Molecular variation of the TYLCV genome
To evaluate the extent of TYLCV variation, the mean pairwise diversity index
π and the Watterson parameter
θ were calculated at all sites along the TYLCV genome by DnaSP Version 5.0 (Fig.
2). Using a sliding window of 100 nts with a 25-step size, we found that while the overall variability for the TYLCV genome was below 3% at the nucleotide level, distribution patterns were uneven. The peak of variability was located within the intergenic region and 5'-terminal part of V2, whereas the most conserved regions were located in the overlapping regions of V1 and V2. Estimation of the average mutation rate showed that the mutation rate of the full-length TYLCV genome reached 1.69×10
−3 subs/site/year (Table
2), which is within the ranges for RNA viruses and EACMV (Jenkins et al.,
2002; Duffy and Holmes,
2009). Further analysis showed that the substitution rate was even higher for the intergenic region, with a substitution rate of 4.81×10
−3 subs/site/year. Rates of nucleotide substitution were also higher in the genes (
C1,
C2,
C3, and
C4) encoded on the complementary strand than those (
V1 and
V2) encoded on the virion strand (Table
2). Statistical tests revealed that the substitution rates estimated for the natural population of TYLCV were significantly higher than those generated from RCA error (1.8×10
−5). These results indicate that the intergenic region had evolved significantly faster than the other ORFs.
Fig.2
Distribution of genetic variation estimated by nucleotide diversity (π) and Watterson’s parameter (θ) for TYLCV
The sliding window size is 100 sites wide, slide by 25-site intervals. The relative positions of the six ORFs and the intergenic region of the TYLCV genome are illustrated by lines above the plot. IR: intergenic region
Table 2
Estimates of average evolutionary divergences and nucleotide substitution rates among TYLCV isolates collected from 2006 to 2010
| Genomic region |
Sequence length (nt) |
Number of sequences |
Genetic distance*
|
Nucleotide substitution rate (substitutions per site per year)
|
| Mean value |
Corrected mean value |
| Full genome |
2781 |
122 |
0.007±0.001 |
1.69×10−3
|
1.67×10−3
|
| IR |
313 |
122 |
0.023±0.005 |
4.81×10−3
|
4.79×10−3
|
|
V1
|
777 |
122 |
0.005±0.002 |
9.52×10−4
|
9.34×10−4
|
|
V2
|
351 |
122 |
0.005±0.002 |
8.50×10−4
|
8.32×10−4
|
|
C1
|
1074 |
122 |
0.005±0.002 |
1.39×10−3
|
1.37×10−3
|
|
C2
|
408 |
122 |
0.009±0.002 |
2.64×10−3
|
2.62×10−3
|
|
C3
|
405 |
122 |
0.008±0.002 |
1.99×10−3
|
1.97×10−3
|
|
C4
|
294 |
122 |
0.005±0.002 |
1.38×10−3
|
1.36×10−3
|
*Genetic distance: the numbers of base substitutions per site from averaging over all sequence pairs; data are shown as mean±standard error (n=100), obtained by a bootstrap procedure
The analyses were conducted using the Kimura’s two-parameter model
To reveal further the process shaping TYLCV evolution, we tested whether specific kinds of mutational changes were overrepresented in the natural population of TYLCV. Our results showed that the shifts from C to T, T to C, G to A, and G to T were overrepresented relative to other kinds of mutational changes. These kinds of mutations constituted 62% of the mutations. However, the transversions of A to C, T to A, and C to G appeared to be underrepresented, constituting only about 10% of the mutations (Table
3). Therefore, these results indicate that transition biases exist in the evolution of TYLCV. These biases favor a shift from CG to AT genome content, especially with a bias in C to T and G to A transitions.
Table 3
Percentages of nucleotide substitutions in TYLCV
| Original base |
Percentage of nucleotide substitution (%)
|
| A |
T |
C |
G |
| A |
|
4.19 |
3.22 |
7.96 |
| T |
3.59 |
|
15.6 |
7.24 |
| C |
4.62 |
26.1 |
|
3.86 |
| G |
9.84 |
10.46 |
3.33 |
3.3. Genetic distances between subpopulations of TYLCV
Genetic distance refers to the average number of nucleotide substitutions between two randomly selected sequences in a population, and can be used to evaluate the genetic variation of a virus population. Using the Kimura’s two-parameter method, the mean genetic distances for the intergenic region and individual ORFs ranged from 0.005 to 0.023. Although the individual ORFs of the TYLCV genome presented similar genetic distances, the mean values for the intergenic region showed the highest genetic distances (Table
2). To understand better the genetic variation of the 26 isolates, the full-length genomic sequences were divided into several subgroups according to the year of collection. Analysis showed that the highest genetic distance was found for the intergenic region (IR) within subgroups (Table
4). Although we did not observe a quasi-linear increase in genetic diversity across years, calculation of the
F
st, the coefficient used to evaluate the extent of genetic differentiation or the gene flow between two populations, showed that the values of
F
st among subgroups 2006 and 2009, 2006 and 2010, as well as 2009 and 2010 were all <0.33, suggesting frequent gene flow (Table
5).
Table 4
Estimates of average evolutionary divergence over sequence pairs within groups arranged by sample collection date
| Genomic region |
Genetic distance within subgroups
|
| 2006 |
2007 |
2008 |
2009 |
2010 |
| IR |
0.007±0.004 |
0.003±0.002 |
0.009±0.002 |
0.018±0.004 |
0.017±0.004 |
|
V1
|
0.003±0.002 |
0.001±0.001 |
0.002±0.001 |
0.005±0.002 |
0.004±0.001 |
|
V2
|
0.002±0.002 |
0.001±0.000 |
0.002±0.001 |
0.004±0.002 |
0.005±0.002 |
|
C1
|
0.004±0.001 |
0.001±0.000 |
0.002±0.000 |
0.004±0.001 |
0.004±0.001 |
|
C2
|
0.008±0.003 |
0.003±0.002 |
0.004±0.001 |
0.005±0.002 |
0.007±0.003 |
|
C3
|
0.006±0.003 |
0.001±0.001 |
0.003±0.001 |
0.006±0.002 |
0.009±0.003 |
|
C4
|
0.003±0.002 |
0.002±0.001 |
0.003±0.001 |
0.004±0.002 |
0.004±0.002 |
Table 5
F
st values for pairs of temporal subpopulations of TYLCV
| Year |
F
st
|
| 2006 |
2007 |
2008 |
2009 |
| 2007 |
0.701 18 |
| 2008 |
0.630 97 |
0.373 27 |
| 2009 |
0.167 67 |
0.606 62 |
0.548 62 |
| 2010 |
0.296 72 |
0.619 52 |
0.569 20 |
0.133 70 |
F
st is a coefficient of the extent of genetic differentiation and provides an estimate of the extent of gene flow. An absolute value of F
st>0.33 suggests infrequent gene flow
3.4. Selection constraints acting on TYLCV genes
Pairwise genetic differences at the synonymous (
d
s) and nonsynonymous (
d
n) nucleotide positions were estimated according to the Pamilo-Bianchi-Li method. The ratio between nucleotide diversity values in nonsynonymous and synonymous positions (
d
n/
d
s) (Table
6) provides an estimation of the degree and direction of the selective constraints acting on the coding regions of TYLCV. Overall, the values of the
d
n/
d
s ratio for the
V1,
V2,
C1, and
C2 genes were markedly low (0.154–0.44), indicating that these genes are under negative selection. However, the ratios of
d
n/
d
s for the
C3 and
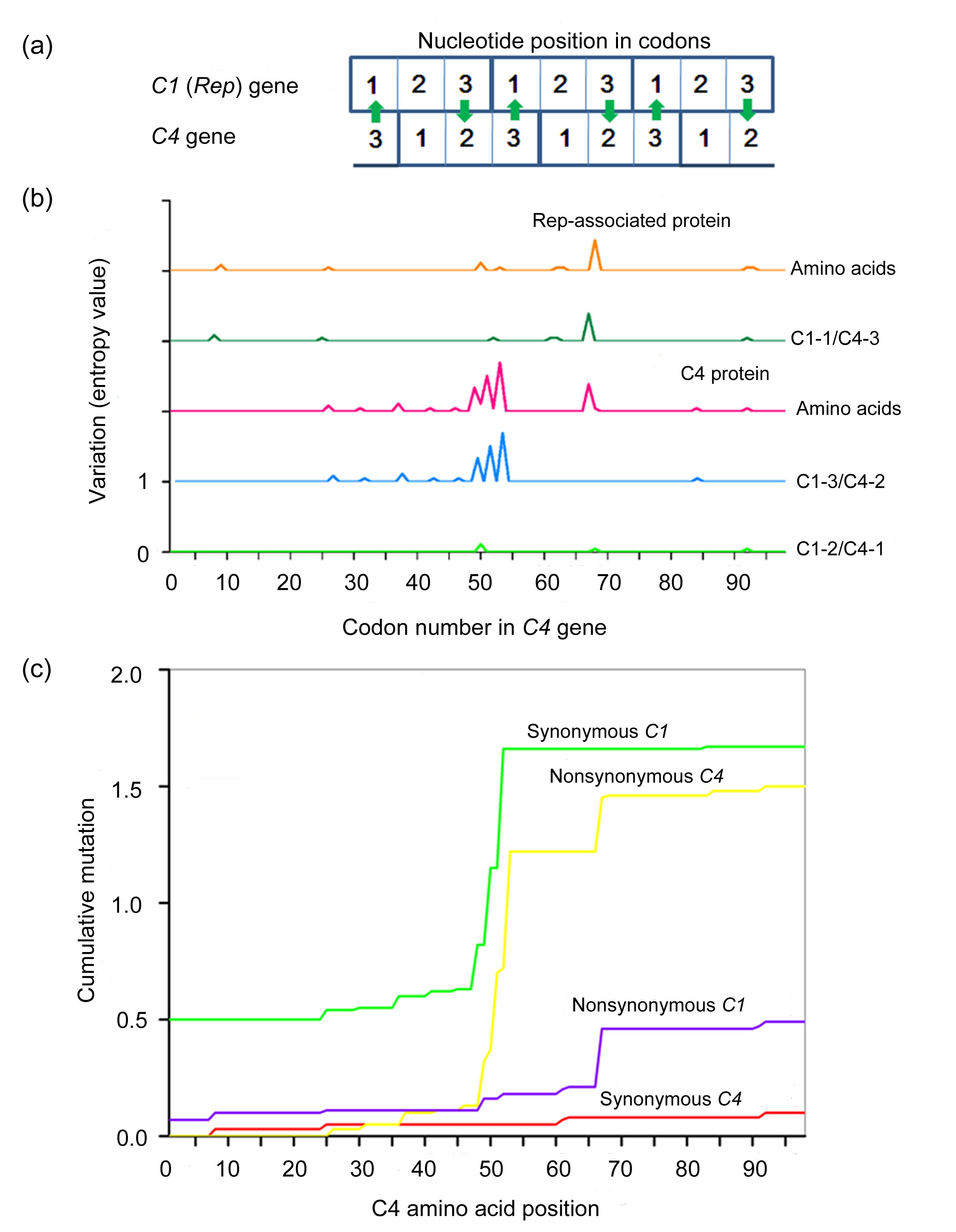
C4 genes were greater than 1. This was probably due to the overlapping regions of C3-C2 and C4-C1, respectively. In the overlapping, frame-shifted C1/C4 region of TYLCV, the first nucleotide position in the C1 codons corresponds to the third position in the C4 codons (C1-1/C4-3), the second position in the C1 codons corresponds to the first position in the C4 codons (C1-2/C4-1), and the third position in the C1 codons corresponds to the second position in the C4 codons (C1-3/C4-2) (Fig.
3a). Independent evolution of overlapping genes has been described for several viruses, with one of the two overlapping genes being subjected to negative selection, while the other is subjected to positive selection. To evaluate the variation between C1 and C4, entropy values for individual amino acids and nucleotides were determined using the BioEdit software. The amino acid changes in the Rep protein were largely determined by nucleotide mutations at the C1-1/C4-3 sites, whereas those in the C4 protein were primarily caused by mutations at the C1-3/C4-2 sites (Fig.
3b). Furthermore, mutations at the C1-2/C4-1 sites, which would affect the amino acid sequences of both the Rep and C4 proteins, were rare. Cumulative synonymous and nonsynonymous mutations plotted using SNAP showed that the
C1 gene accumulated more synonymous mutations, while the
C4 gene accumulated more nonsynonymous mutations (Fig.
3c). Taken together, these results revealed that the overlapping regions of
C1 and
C4 are under evolutionary constraints, allowing for more rapid evolution of the C4 protein while minimizing amino acid changes in the conserved Rep protein.
Table 6
Estimation of nucleotide diversity of different genes in TYLCV
| Gene |
d
n
|
d
s
|
d
n/d
s
|
|
V1
|
0.002±0.001 |
0.013±0.004 |
0.154 |
|
V2
|
0.002±0.002 |
0.010±0.005 |
0.200 |
|
C1
|
0.004±0.001 |
0.009±0.004 |
0.440 |
|
C2
|
0.006±0.003 |
0.023±0.013 |
0.260 |
|
C3
|
0.009±0.004 |
0.004±0.004 |
2.250 |
|
C4
|
0.008±0.003 |
0.002±0.001 |
4.000 |
d
n: nonsynonymous substitution; d
s: synonymous substitution.d
n and d
s are expressed as mean±standard error
Fig.3
Variation in nucleotides and amino acids of the overlapping C1 and C4 genes
(a) Frame-shifted positions of the overlapping C1 and C4 genes. Mutations in the open reading frames of the overlapping C1 and C4 genes result in differential rates of amino acid changes. For example, a nucleotide mutation in the third position of the C1 codon is likely to be synonymous, allowing a nonsynonymous mutation in the C4 codon (arrows). (b) Levels of nucleotide and amino acid variation in the three sets of nucleotides in relation to the nucleotide positions of the codons in the overlapping Rep and C4 proteins. (c) Cumulative incidences of synonymous and nonsynonymous mutations in the overlapping region of C1/C4. The x axis represents the position of the codon in C4, and the y axis represents the cumulative values of synonymous or nonsynonymous mutations estimated at a specific codon position
4. Discussion
Before 2006, no begomovirus or TYLCV had been detected in Shanghai. Due to the rapid introduction and severe outbreak of the Q biotype of whitefly in China, TYLCV has now moved to several provinces of China, causing unprecedented economic losses, especially in tomato plants. Prior to this field survey, no other competitive begomovirus had been described in Shanghai. This epidemiological situation provides a great opportunity to investigate the genetic structure and evolution of TYLCV following its introduction into a new region.
This study analyzed the genetic variability of 26 TYLCV isolates collected from Shanghai over a five-year period. Analysis of the full-length genomic DNA sequences revealed that the level of genetic variation observed in the natural population of TYLCV (1.69×10
−3 subs/site/year) was similar to that found in RNA viruses (Duffy et al.,
2008; Simmons et al.,
2008; Pagan and Holmes,
2010; Pagan et al.,
2010). These and other results suggest that a considerable amount of sequence variation occurs within and between populations of geminiviruses, challenging the view that RNA viruses are prone to higher mutation rates than DNA viruses (Domingo and Holland,
1997; Roossinck,
1997; Isnard et al.,
1998; Sanz et al.,
1999). It has been speculated that the high mutation rate of geminivirus genomes is in part due to improper methylation patterns occurring during their replication (Ge et al.,
2007; Duffy and Holmes,
2008). However, recent studies have shown that geminivirus genomes are subjected to DNA methylation in infected host plants (Raja et al.,
2008;
2010; Yang et al.,
2011; Zhang et al.,
2011). Thus, it is possible that base-excision repair may not act on geminivirus genomes because the double-stranded state lasts for a very short time during rolling circle replication. The high mutation rate is also probably in part due to base deamination occurring spontaneously or by the action of deaminating host enzymes (Duffy and Holmes,
2008; van der Walt et al.,
2008). This may be supported by the observation of C to T transition biases in this study. Indeed, previous studies have also revealed the same transition biases as our genomic analysis (Ge et al.,
2007; Duffy and Holmes,
2008;
2009).
Although we did not observe a significant quasi-linear increase in genetic diversity across years, we found frequent gene flow between isolates obtained from the years 2006, 2009, and 2010. As was reported for TYLCV surveyed in an insular environment, the amount of mutations analyzed in this study was greater than that found in tomato yellow leaf curl Sardinia virus (TYLCSV) over an eight-year period, using a single-strand conformation polymorphism technique (Sanchez-Campos et al.,
2002; Delatte et al.,
2007). This discrepancy might be explained by the different techniques used. The RCA technique used in this study has revolutionized the diagnosis and genomics of geminiviruses, allowing for a more efficient and reliable way to obtain full-length geminivirus DNA (Haible et al.,
2006). Our results also indicated that the mutation rate detected in the natural TYLCV population was greater than that caused by phi29 DNA polymerase and sequencing errors. Alternatively, this discrepancy might also come from the viruses used. It seems that TYLCV causes more severe damage to crop production, as surveys conducted between 1996 and 1998 in Málaga showed a progressive displacement of TYLCSV by TYLCV (Sanchez-Campos et al.,
1999). Indeed, since the first introduction of TYLCV into China, it has become the most prevalent begomovirus in China.
Estimation of nucleotide diversities using the ratio between nonsynonymous and synonymous positions (
d
n/
d
s) suggested that V1, V2, C1, and C2 were more tolerant to amino acid changes than C3 and C4. Among all the coding regions analyzed, the
C4 gene embedded in the
C1 gene appeared to be the most flexible and might be subjected to positive selection. We demonstrated that the evolution of the overlapping
C1 and
C4 genes occurs primarily via C1-1/C4-3 and C1-3/C4-2 sites, respectively, and mutations at the C1-2/C4-1 sites, which were most likely to alter the amino acid sequences of both the Rep and C1 proteins, were rare. Further SNAP analysis showed that positions in the C-terminal of the
C4 gene, which were under positive selection, were subjected to purifying selection in the overlapping region of the
C1 gene. Indeed, a similar phenomenon has been demonstrated for the evolution of tomato leaf deformation virus, an indigenous new world (NW) monopartite begomovirus emerging from the DNA-A component of an NW bipartite progenitor (Melgarejo et al.,
2013). We speculated that this would be correlated with the function of the C4 protein. Compared to the conserved Rep protein, the functions of the C4 protein are more divergent among monopartite and bipartite begomoviruses. For monopartite begomoviruses, C4 is a pathogenicity determinant that is involved in movement and suppression of host defense responses, whereas the homologues of C4 encoded by most NW begomoviruses do not play a role in pathogenicity. This strategy, which allows viruses to condense a maximal amount of information into short genomes, has been widely used by a number of animal and plant-infecting viruses (Zaaijer et al.,
2007). Given the rapid spread of TYLCV in China, it will be interesting to monitor the evolutionary constraints
in vivo in experimentally infected tomato plants.
* Project supported by the Special Fund for Agro-Scientific Research in the Public Interest from the Ministry of Agriculture, China (No. 201003065), the China Postdoctoral Science Foundation (No. 2012M510200), and the National Natural Science Foundation of China (No. 31200117)Compliance with ethics guidelines Xiu-ling YANG, Meng-ning ZHOU, Ya-juan QIAN, Yan XIE, and Xue-ping ZHOU declare that they have no conflict of interest.References
[1] Cohen, S., Antignus, Y., 1994. Tomato yellow leaf curl virus, a whitefly-borne geminivirus of tomatoes. Advances in Disease Vector Research. Springer-Verlag,New York :259-288.

[2] Czosnek, H., Ghanim, M., 2011.
Bemisia tabaci—tomato yellow leaf curl virus interaction causing worldwide epidemics.
The Whitefly, (Homoptera: Aleyrodidae) Interaction with Geminivirus-Infected Host Plants, Springer Netherlands,:51-67.
[3] Delatte, H., Holota, H., Moury, B., 2007. Evidence for a founder effect after introduction of tomato yellow leaf curl virus-mild in an insular environment.
J Mol Evol, 65(1):112-118.

[4] Domingo, E., Holland, J.J., 1997. RNA virus mutations and fitness for survival.
Annu Rev Microbiol, 51(1):151-178.
[5] Duffy, S., Holmes, E.C., 2008. Phylogenetic evidence for rapid rates of molecular evolution in the single-stranded DNA begomovirus tomato yellow leaf curl virus.
J Virol, 82(2):957-965.
[6] Duffy, S., Holmes, E.C., 2009. Validation of high rates of nucleotide substitution in geminiviruses: phylogenetic evidence from East African cassava mosaic viruses.
J Gen Virol, 90(6):1539-1547.
[7] Duffy, S., Shackelton, L.A., Holmes, E.C., 2008. Rates of evolutionary change in viruses: patterns and determinants.
Nat Rev Genet, 9(4):267-276.
[8] Ge, L., Zhang, J., Zhou, X., 2007. Genetic structure and population variability of tomato yellow leaf curl China virus.
J Virol, 81(11):5902-5907.
[9] Glick, E., Zrachya, A., Levy, Y., 2008. Interaction with host SGS3 is required for suppression of RNA silencing by tomato yellow leaf curl virus V2 protein.
PNAS, 105(1):157-161.
[10] Guo, W., Jiang, T., Zhang, X., 2008. Molecular variation of satellite DNAβ molecules associated with
Malvastrum yellow vein virus and their role in pathogenicity.
Appl Environ Microbiol, 74(6):1909-1913.
[11] Guo, W., Yang, X., Xie, Y., 2009. Tomato yellow leaf curl Thailand virus-[Y72] from Yunnan is a monopartite begomovirus associated with DNAβ.
Virus Genes, 38(2):328-333.
[12] Haible, D., Kober, S., Jeske, H., 2006. Rolling circle amplification revolutionizes diagnosis and genomics of geminiviruses.
J Virol Methods, 135(1):9-16.
[13] Hall, T., 1999. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT.
Nucl Acids Symp Ser, 41:95-98.
[14] Hanley-Bowdoin, L., Settlage, S.B., Orozco, B.M., 1999. Geminiviruses: models for plant DNA replication, transcription, and cell cycle regulation.
Crit Rev Biochem Mol Biol, 18(1):71-106.
[15] Isnard, M., Granier, M., Frutos, R., 1998. Quasispecies nature of three maize streak virus isolates obtained through different modes of selection from a population used to assess response to infection of maize cultivars.
J Gen Virol, 79(12):3091-3099.
[16] Jenkins, G.M., Rambaut, A., Pybus, O.G., 2002. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis.
J Mol Evol, 54(2):156-165.
[17] Jiang, L., Wei, C.H., Li, Y., 2012. Viral suppression of RNA silencing.
Sci China C Life Sci, 55(2):109-118.
[18] Korber, B., 2002. HIV signature and sequence similarities. Computational and Evolutionary Analysis of HIV Molecular Sequences. Kluwer Academic Publishers,Boston, Massachusetts :55-72.
[19] Librado, P., Rozas, J., 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data.
Bioinformatics, 25(11):1451-1452.
[20] Melgarejo, T.A., Kon, T., Rojas, M.R., 2013. Characterization of a new world monopartite begomovirus causing leaf curl disease of tomato in Ecuador and Peru reveals a new direction in geminivirus evolution.
J Virol, 87(10):5397-5413.
[21] Moriones, E., Navas-Castillo, J., 2000. Tomato yellow leaf curl virus, an emerging virus complex causing epidemics worldwide.
Virus Res, 71(1-2):123-134.
[22] Padidam, M., Beachy, R.N., Fauquet, C.M., 1995. Classification and identification of geminiviruses using sequence comparisons.
J Gen Virol, 76(Pt 2):249-263.
[23] Pagan, I., Holmes, E.C., 2010. Long-term evolution of the
Luteoviridae: time scale and mode of virus speciation.
J Virol, 84(12):6177-6187.
[24] Pagan, I., Firth, C., Holmes, E.C., 2010. Phylogenetic analysis reveals rapid evolutionary dynamics in the plant RNA virus genus tobamovirus.
J Mol Evol, 71(4):298-307.
[25] Raja, P., Sanville, B.C., Buchmann, R.C., 2008. Viral genome methylation as an epigenetic defense against geminiviruses.
J Virol, 82(18):8997-9007.
[26] Raja, P., Wolf, J.N., Bisaro, D.M., 2010. RNA silencing directed against geminiviruses: post-transcriptional and epigenetic components.
Biochim Biophys Acta, 1799(3):337-351.
[27] Roossinck, M.J., 1997. Mechanisms of plant virus evolution.
Annu Rev Phytopathol, 35(1):191-209.
[28] Sanchez-Campos, S., Navas-Castillo, J., Camero, R., 1999. Displacement of tomato yellow leaf curl virus (TYLCV)-Sr by TYLCV-Is in tomato epidemics in Spain.
Phytopathology, 89(11):1038-1043.
[29] Sanchez-Campos, S., Diaz, J.A., Monci, F., 2002. High genetic stability of the
Begomovirus tomato yellow leaf curl
Sardinia virus in southern Spain over an 8-year period.
Phytopathology, 92(8):842-849.
[30] Sanz, A.I., Fraile, A., Gallego, J.M., 1999. Genetic variability of natural populations of cotton leaf curl geminivirus, a single-stranded DNA virus.
J Mol Evol, 49(5):672-681.
[31] Simmons, H.E., Holmes, E.C., Stephenson, A.G., 2008. Rapid evolutionary dynamics of zucchini yellow mosaic virus.
J Gen Virol, 89(4):1081-1085.
[32] Tamura, K., Peterson, D., Peterson, N., 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods.
Mol Biol Evol, 28(10):2731-2739.
[33] Thompson, J.D., Gibson, T.J., Higgins, D.G., 2002. Multiple sequence alignment using ClustalW and ClustalX.
Curr Prot Bioinf, 00:2.3.1-2.3.22.
[34] van der Walt, E., Martin, D.P., Varsani, A., 2008. Experimental observations of rapid Maize streak virus evolution reveal a strand-specific nucleotide substitution bias.
Virol J, 5:104
[35] Wu, J.B., Dai, F.M., Zhou, X.P., 2006. First report of tomato yellow leaf curl virus in China.
Plant Dis, 90(10):1359-1359.
[36] Yang, X., Xie, Y., Raja, P., 2011. Suppression of methylation-mediated transcriptional gene silencing by βC1-SAHH protein interaction during geminivirus-betasatellite infection.
PLoS Pathog, 7(10):e1002329
[37] Zaaijer, H.L., van Hemert, F.J., Koppelman, M.H., 2007. Independent evolution of overlapping polymerase and surface protein genes of hepatitis B virus.
J Gen Virol, 88(Pt 8):2137-2143.
[38] Zhang, H., Gong, H., Zhou, X., 2009. Molecular characterization and pathogenicity of tomato yellow leaf curl virus in China.
Virus Genes, 39(2):249-255.
[39] Zhang, Z., Chen, H., Huang, X., 2011. BSCTV C2 attenuates the degradation of SAMDC1 to suppress DNA methylation-mediated gene silencing in
Arabidopsis
.
Plant Cell, 23(1):273-288.
[40] Zhou, X.P., Xie, Y., Zhang, Z.K., 2001. Molecular characterization of a distinct begomovirus infecting tobacco in Yunnan, China.
Arch Virol, 146(8):1599-1606.
Open peer comments: Debate/Discuss/Question/Opinion
<1>